Diese Veränderungen führen zu einer verstärkten Dehnung des Körpergewebes, was die Ursache für alle Erscheinungsformen der Krankheit ist.

- Thoraxdeformitäten bei Kindern und Jugendlichen – ein umfassendes Konzept für die Behandlung angeborener und erworbener Deformitäten
- Angeborene Thoraxdeformitäten bei Kindern
- Marfan-Syndrom
- Sporadische (nicht erbliche) Deformitäten
- Marfan-Syndrom – was sind seine Merkmale? Art der Vererbung.
- Symptome der Krankheit
- Diagnose des Marfan-Syndroms
- Wie wird der Test durchgeführt?
- BEHANDLUNG UND VORBEUGUNG
- Das Aussehen eines Kindes mit Marfan-Syndrom.
- Beispiele für ähnliche Studien
- Management von Schwangerschaft und Geburt bei einigen Arten von extragenitalen Pathologien
- Behandlung und Vorbeugung von rheumatischen
- Achondroplasie
- Duchenne-Myodystrophie
- Arten von Erbkrankheiten
- Chromosomale Krankheiten
- Monogenetische Krankheiten
- Mitochondriale Krankheiten
- Wie Erbkrankheiten vererbt werden
- Autosomal-dominantes Vererbungsmuster
- Autosomal rezessives Vererbungsmuster
- Klassische Symptome der Marfan-Krankheit
- Korrektur und Behandlungsmethoden
- Marfan-Syndrom – Arten
- Marfan-Syndrom – Diagnose in Israel
- Diagnose des Marfan-Syndroms in Israel
- Der erste Tag
- Zweiter Tag
- Tag drei
- Wie hoch sind die Kosten für die Behandlung des Marfan-Syndroms in Israel?
Thoraxdeformitäten bei Kindern und Jugendlichen – ein umfassendes Konzept für die Behandlung angeborener und erworbener Deformitäten
Das Turner Children’s Traumatology and Orthopaedic Research Centre bietet ein umfassendes Konzept für die Behandlung angeborener und erworbener Deformitäten. Es werden bedeutende Forschungsarbeiten zur Diagnose und Behandlung von angeborenen und erworbenen Erkrankungen des Bewegungsapparats bei Kindern durchgeführt. Insbesondere wurden fortschrittliche Methoden zur Behandlung von angeborenen und erworbenen Thoraxdeformitäten erfolgreich in der ersten Neugeborenenintensivstation eingeführt. Dank der wissenschaftlichen und klinischen Aktivitäten unserer Spezialisten entwickelt sich dieser Bereich aktiv weiter.
Für jeden Fall wird ein individueller Therapieansatz entwickelt. Die Behandlung erfolgt konservativ oder chirurgisch, je nach dem Zustand des einzelnen Patienten. Die chirurgische Behandlung von Thoraxdeformitäten erfolgt im Rahmen einer spezialisierten und hochmodernen medizinischen Versorgung (VMP im Rahmen des Systems der obligatorischen Krankenversicherung für alle russischen Bürger, für ausländische Bürger auf Honorarbasis). Die präoperative Untersuchung und Behandlung wird für Patienten aus allen Regionen Russlands auf Kosten des Staates durchgeführt.
Eine Thoraxdeformität ist eine angeborene oder erworbene Veränderung des muskuloskelettalen Skeletts und der Form des Brustkorbs.
Die Frage, was die Ursache für eine Thoraxdeformität bei einem Kind ist, beschäftigt alle Eltern. Alle Verformungen sind entweder angeboren oder erworben.
Angeborene Thoraxdeformitäten bei Kindern
Angeborene Thoraxdeformitäten bei Kindern können genetisch bedingt sein, aber auch durch Veränderungen bei der Bildung des Sternoklavikularkomplexes, die zu einer allmählichen Zunahme der Deformitäten führen können, bis das Skelett des Kindes oder Jugendlichen vollständig ist.
Angeborene Fehlbildungen sind mit einer abnormen Entwicklung des Skeletts (Wirbelsäule, Rippen) verbunden, die auf ein Ungleichgewicht im Mineral- und Hormonhaushalt zurückzuführen ist. Die Folgen davon können eine spezifische Entwicklung des Körpers sein:
- schmerzhafte Schlankheit;
- schmale Schultern;
- hohe Körpergröße;
- hervortretende Schulterblätter und Schlüsselbeine;
- hohle Brust beim Einatmen;
- lange Gliedmaßen;
- Krümmungen der Wirbelsäule (Skoliose oder Kyphose).
Bei 20-65 % der Thoraxdeformitäten werden erbliche Deformitäten diagnostiziert. Es gibt Krankheiten und spezifische Syndrome, bei denen diese Art von Deformität eines der Symptome ist. So ist es zum Beispiel nicht ungewöhnlich, dass sich diese Pathologie vor dem Hintergrund eines Marfan-Syndroms entwickelt.
Marfan-Syndrom
Diese Erkrankung ist durch eine trichterförmige und kielförmige Verformung des Brustkorbs gekennzeichnet.
Das Marfan-Syndrom ist durch die folgenden Symptome gekennzeichnet
- asthenischer Körperbau;
- Arachnodaktylie;
- dissezierendes Aortenaneurysma;
- Verlagerung oder Subluxation der Augenlinse (oder andere Sehstörungen);
- biochemische Veränderungen des Kollagen- und Glykosaminoglykan-Stoffwechsels.
Bindegewebs- und Knorpeldysplasien, die durch enzymatische Anomalien verursacht werden, können zur Entwicklung von Thoraxdeformitäten beitragen.
Sporadische (nicht erbliche) Deformitäten
Nicht erbliche Thoraxdeformitäten entstehen durch teratogene Faktoren, die den Fötus während seiner Entwicklung beeinträchtigen. Am häufigsten werden die Fehlbildungen durch ein asynchrones, nicht harmonisches Wachstum des Brustbeins und der Rippenknorpel verursacht.
Marfan-Syndrom – was sind seine Merkmale? Art der Vererbung.

Die Krankheit wird autosomal dominant vererbt. Sie wird durch Mutationen im FBN1-Gen verursacht, das die Synthese von Fibrillin steuert, einem der wichtigsten Proteine für das normale Leben, das die Elastizität des Bindegewebes erhält. Beim Morbus Marfan liegt ein Mangel an diesem Protein vor, wodurch das Bindegewebe an Festigkeit und Elastizität verliert und die Betroffenen nicht in der Lage sind, hohen physiologischen Belastungen standzuhalten. Auch die Aorta und das Zimtband des Auges, die große Mengen an Fibrillin enthalten, sind geschädigt. Elastische Fasern sind im gesamten menschlichen Körper in großer Zahl vorhanden, aber in diesen Strukturen sind sie am stärksten konzentriert. Es ist nicht ungewöhnlich, dass ein Aortenaneurysma zum Tod führt.
Die medizinische Forschung bestätigt, dass 75 Prozent der Fälle des Marfan-Syndroms familiär bedingt sind, während nur die restlichen 25 Prozent auf eine sporadische Primärmutation zurückzuführen sind.
Es wurde auch festgestellt, dass die Wahrscheinlichkeit, ein Kind mit Marfan-Syndrom zu bekommen, mit dem Alter des Vaters steigt: je älter der Vater, desto höher das Risiko.
Symptome der Krankheit
Das Marfan-Syndrom geht mit einer Reihe von Anomalien einher. Die sogenannte ‚Marfan-Trias‘ umfasst Schäden an den Skelettknochen, Herz-Kreislauf-Erkrankungen und verschiedene Sehstörungen. Die geistige Leistungsfähigkeit ist von diesem Syndrom jedoch nicht betroffen.
Die Patienten neigen zu einem unproportionalen Körperbau: bei relativ großer Körpergröße haben sie einen verkürzten Rumpf, lange Arme und spinnenartige Finger (Arachnodaktylie), einen asthenischen Körperbau mit Muskelhypotonie, Thoraxdeformitäten, schwere Wirbelsäulenverkrümmungen (Kyphose, Skoliose, zervikale Subluxationen und Dislokationen) und Plattfüße.
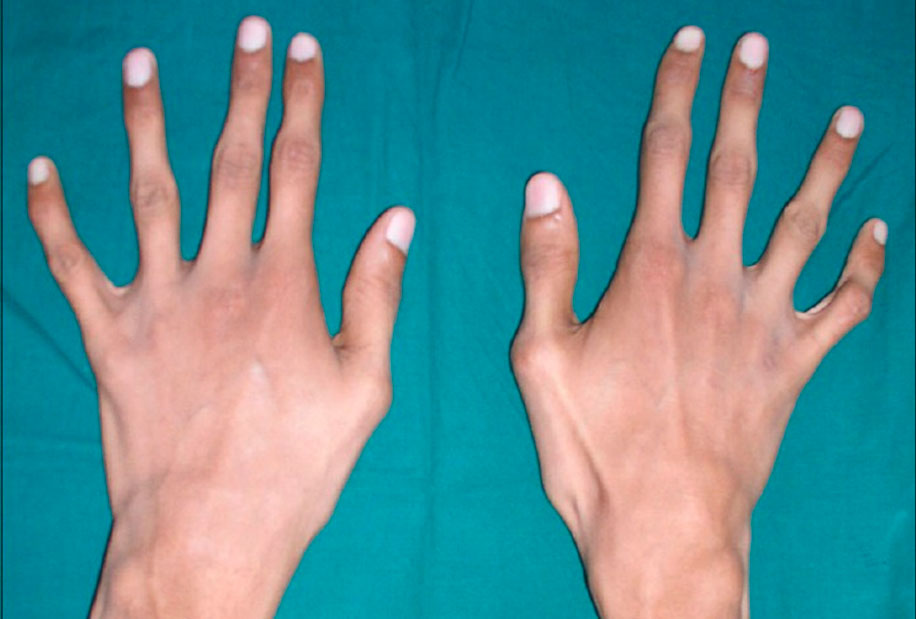
Das Marfan-Syndrom, das mit Anomalien des Kreislaufsystems einhergeht, äußert sich durch Defekte an den Wänden der großen Gefäße, insbesondere der Aorta und der großen Äste der Lungengefäße, sowie durch Fehlbildungen des Herzens. Diese Läsionen bilden sich oft schon beim sich entwickelnden Fötus. Die schwerste Form des Marfan-Syndroms, bei der die Symptome bereits bei der Geburt auftreten, führt zu fortschreitendem Herzversagen und zum Tod im ersten Lebensjahr des Kindes.
Diagnose des Marfan-Syndroms
Die wichtigste Methode zur Diagnose von Herz-Kreislauf-Schäden ist die EchoCG-Studie.
Risikofaktoren für eine Aortendissektion beim Marfan-Syndrom:
- Aortendurchmesser >5 cm;
- Dilatation, die über den Sinus Valsalva hinausgeht;
- Schnell fortschreitende Dilatation (>5% oder 2 mm und 1 Jahr bei Erwachsenen);
- Familienanamnese einer Aortendissektion.
Bei allen Patienten mit Marfan-Syndrom sollte jährlich eine klinische Untersuchung und ein transthorakales EchoCG durchgeführt werden. Bei Kindern wird das EchoCG in Abhängigkeit vom Durchmesser und der Geschwindigkeit der Aortendilatation eingesetzt. Bei schwangeren Frauen mit Marfan-Syndrom besteht das Risiko einer Aortendissektion, wenn der Aortendurchmesser 4 cm überschreitet. In solchen Fällen ist eine Überwachung der kardiovaskulären Funktion während der Schwangerschaft und der Entbindung angezeigt.
Die Diagnose einer fortschreitenden Aortendilatation wird durch das Vorhandensein von Oxyprolin und Glykosaminoglykanen im täglichen Urin gestellt, wobei die Ausscheidung um das 2-3fache ansteigt.
[10], [11], [12], [13], [14]
Wie wird der Test durchgeführt?
Starke körperliche Betätigung ist kontraindiziert. Massagen und physiotherapeutische Behandlungen sind bereits im Kindesalter angezeigt. Chirurgische Behandlung von Augenpathologien, Herzklappen und Aneurysmen. Das Risiko einer Aortendissektion bei Patienten mit Marfan-Syndrom kann durch den Einsatz von Betablockern zur Senkung des systolischen Aortendrucks verringert werden, was zur Grundlage klinischer Leitlinien für die Behandlung von Patienten mit Marfan-Syndrom geworden ist:
- Bei Patienten aller Altersgruppen mit Aortendilatation wird die größte prophylaktische Wirkung von Betablockern beim Aortendurchmesser beobachtet
Das iLive-Portal bietet keine medizinische Beratung, Diagnose oder Behandlung.
Die auf dem Portal veröffentlichten Informationen dienen lediglich der Orientierung und sollten nicht ohne Rücksprache mit einem Fachmann verwendet werden.
Bitte lesen Sie die Regeln und Richtlinien des Portals sorgfältig durch. Sie können auch Kontakt mit uns aufnehmen!
Urheberrecht © 2011 – 2023 iLive. Alle Rechte vorbehalten.
Klicken Sie einfach auf die Schaltfläche ‚Anfrage senden‘, um uns Informationen zukommen zu lassen. Sie können auch einen Kommentar hinzufügen.
BEHANDLUNG UND VORBEUGUNG
Es gibt keine spezifische Behandlung für diese Krankheit: Es ist nicht möglich, die Gene vor der Geburt zu verändern. Die Behandlung erfolgt nur symptomatisch und hängt von den körperlichen Veränderungen ab, die sich bei einem Patienten mit Marfan-Syndrom entwickeln . Einige Komplikationen der Pathologie können erfolgreich korrigiert und andere chirurgisch behandelt werden.
Der Patient muss von einem Team von Fachärzten – Augenarzt, Neurologe, Kardiologe, Orthopäde und Chirurg – untersucht werden. Das Hauptziel der Therapie besteht darin, die Funktion des Herzens und der Blutgefäße zu unterstützen.
(Adrenoblocker, Antiarrhythmika, Antikoagulantien, usw.).
(Herzklappenfehlfunktion, Dilatation, Lungenarteriendissektion), Aorta, Klappenprothesen.
Normalisierung des Sehvermögens wird erreicht durch Korrektur der Kurzsichtigkeit (Tragen von Brillen, Linsen), Behandlung von grauem und grünem Star, Implantation einer Kunstlinse.
Bei Verletzungen der Gelenke und der Wirbelsäule chirurgische Behandlung (Prothesen, Gelenkplastiken, Beseitigung von Zwischenwirbelbrüchen), Korrektur von Kyphose und Skoliose durch Traktion und manuelle Therapie. Zu den verwendeten Medikamenten gehören Muskelrelaxantien und B-Vitamine. Auch Physiotherapie und physikalische Therapie kommen zum Einsatz.
Wenn die Lunge betroffen ist Wenn die Lunge betroffen ist, ist häufig eine chirurgische Behandlung (Lungendrainage) erforderlich.
Eine Schwangerschaft bei Patienten mit Marfan-Syndrom sollte streng geplant werden und unter der Aufsicht eines auf die Behandlung von Menschen mit Marfan-Syndrom spezialisierten Ärzteteams entwickelt werden. Die Entbindung erfolgt ausschließlich per Kaiserschnitt. Schon vor der Schwangerschaft ist es ratsam, das mögliche Fortschreiten der Aortendissektion zu überprüfen und, wenn möglich, einen Teil des Gefäßes chirurgisch zu ersetzen. Nach Rücksprache mit einem Genetiker kann das ungefähre Risiko der Vererbung der Krankheit berechnet werden.
Bei der Überwachung von Patienten mit Marfan-Syndrom sollten die folgenden Anforderungen an Arbeit, Ruhe und Rehabilitation eingehalten werden:
Das Aussehen eines Kindes mit Marfan-Syndrom.
[Elektronische Quelle]//URL: https://psystars.ru/referat/sindrom-marfana/
1 Zasukhina T.D., Lvova T.N., Vasilyeva I.T. Reduzierte Fähigkeit zur Reparatur von durch Mutagene induzierten DNA-Schäden in Zellen von Patienten mit Marfan-Syndrom. // Berichte der Akademie der Wissenschaften der UdSSR, 1982 – 265 – 5 – pp. 1261 – 1263.
2. lisichenko O.F. Marfan-Syndrom. / Novosibirsk, Nauka, 1986. – 163 с.
3. prozorovskaya N.N., Glinyannaya S.V., Gerashchenko L.P. et al. Wirkung einer Betablocker- und Vitaminkomplextherapie auf die Oxyprolinausscheidung bei einigen erblichen Bindegewebserkrankungen // Problems of Medical Chemistry, 1988 – т. 35. – № 5. – с. 99 – 104
4 Bubnova N.I. Morbus Marfan bei der schwangeren Frau und beim Fötus // Archives of Pathology, 1986 – № 9. – с. 59 – 61. 11. Robert S. Porter, hrsg. von I. I. Dedov. I. Dedov, ‚Leitlinien für die Medizin. Diagnose und Behandlung‘-M, 2015.
5 N.P. Bochkov. Klinische Genetik:Lehrbuch,3. überarbeitete und ergänzte Auflage-Moskau: GEOTAR-MED,2004. S.170-173.
6 N.P. Shabalov. Kinderkrankheiten: Lehrbuch, 5. Aufl., Bd.2-SPb: Peter,2002. pp.482-484.
7. Semjatschkina A.N., Selverova N.B., Lyubchenko L.N.. Endokrine Störungen bei Morbus Marfan. In der Sammlung: Hereditäre Störungen des Wachstums und der Entwicklung bei Kindern. Moskau 1993, S. 55-63.
8. Yakovlev V.M., Dubiley G.S. Restorative treatment in connective tissue dysplasia. Omsk: Izd vo OGMA 1996. 120с.
9. Kadurina T.I. Vererbte Kollagenopathien (Klinik, Diagnose, Behandlung und Nachsorge).
Beispiele für ähnliche Studien
Management von Schwangerschaft und Geburt bei einigen Arten von extragenitalen Pathologien
Merkmale von hämodynamischen Veränderungen. Es werden folgende Arten der neurozirkulatorischen Dystonie unterschieden: kardiale hypotensive hypertensive 1.2 Behandlung von Schwangerschaft und Geburt bei Herzerkrankungen und geburtshilflichen Pathologien. Kontraindikationen für Kaiserschnittoperationen .
Behandlung und Vorbeugung von rheumatischen
. Läsionen sind kasuistisch geworden. 2 Behandlung und Vorbeugung von Rheuma Die Behandlung von Rheuma basiert auf den aktuellen Kenntnissen über die Ätiologie und . bei prädisponierten Personen, hauptsächlich im Alter von 7-15 Jahren. Obwohl es sich nicht um eine Massenerkrankung handelt, ist Rheuma .
Achondroplasie
Die Achondroplasie (Chondrodystrophie) wird durch eine Mutation im Fibroblasten-Wachstumsfaktor-Rezeptor-Gen verursacht, die eine abnorme Aktivität bestimmter Enzyme hervorruft, was zu einem abnormen Wachstum und einer abnormen Entwicklung des Knorpels in den Epiphysen der Röhrenknochen und der Schädelbasis führt.
Die Krankheit ist gekennzeichnet durch eine kurze Statur bei normaler Rumpflänge, einen großen Schädel mit vorstehendem Hinterkopf und einen eingefallenen Nasenrücken. Die Gliedmaßen sind kurz, vor allem proximal des Oberschenkels und des Oberarmknochens, und die Hände sind breit und kurz. Die Kinder sind in ihrer motorischen Entwicklung verzögert; der Intellekt ist normalerweise nicht beeinträchtigt.
Duchenne-Myodystrophie
Duchenne-Myodystrophie – Eine schwere Erkrankung, die durch Muskelschwäche und erhöhte Kreatinphosphokinasewerte im Plasma (10-100fach) gekennzeichnet ist.
Der frühe Beginn im Alter von 3 bis 5 Jahren ist gekennzeichnet durch eine zunehmende Schwäche der Oberschenkel- und Beckenmuskulatur und eine allmähliche Zunahme der Wadenmuskulatur, des Schultergürtels, des Rückens, des Bauches usw. Es kommt zu einem entenartigen Gang. Die Krankheit schreitet stetig voran, und Kinder im Alter von 10-11 Jahren sind bettlägerig. Es wird eine Pseudohypertrophie der Waden- und Gesäßmuskulatur beobachtet, da das Muskelgewebe durch Binde- und Fettgewebe ersetzt wird. In vielen Fällen kommt es aufgrund der Muskelatrophie zu einer Beugekontraktur der Oberschenkel- und Kniegelenke sowie der Gelenke der oberen Gliedmaßen. Die tiefen Sehnenreflexe nehmen frühzeitig ab. Es besteht eine Tendenz zu einer gewissen Abnahme der geistigen Leistungsfähigkeit. Die Lebenserwartung der Patienten liegt bei 20-35 Jahren.
Arten von Erbkrankheiten
Erbkrankheiten werden in Chromosomen-, Gen- und Mitochondrienerkrankungen unterteilt.
Chromosomale Krankheiten
Chromosomenkrankheiten werden durch Veränderungen der Anzahl oder Struktur der Chromosomen verursacht. Sie sind durch gemeinsame Merkmale gekennzeichnet: niedriges Geburtsgewicht und geringe Körperlänge, geistige und körperliche Retardierung, verzögerte und abnorme sexuelle Entwicklung usw.
Chromosomenkrankheiten werden selten vererbt. In mehr als 95 % der Fälle ist das Risiko, dass in der Familie erneut ein Kind mit Chromosomenanomalien geboren wird, nicht höher als in der Allgemeinbevölkerung.
Zu den Chromosomenkrankheiten mit abnormaler Chromosomenzahl gehören:
Chromosomenkrankheiten mit Chromosomenanomalien – Di Giorgi-Syndrom, Wolff-Hirschhorn-Syndrom, Kitts-Syndrom, Alfie-Syndrom, Orbeli-Syndrom.
Monogenetische Krankheiten
Monogene Krankheiten entstehen durch DNA-Schäden auf der Ebene der Gene. Die Zahl der monogenen Krankheiten wird auf bis zu 5.000 geschätzt.
Zu den Merkmalen monogener Krankheiten gehören:
- Verschiedene Formen der geistigen Retardierung,
- Defekte des Gehörs, des Sehvermögens,
- Dysplasie des Skelettsystems,
- Erkrankungen des Nervensystems, des Hormonsystems, des Immunsystems und anderer Systeme.
Einige der bekanntesten monogenen Krankheiten sind. Mukoviszidose, Hämophilie A und B, Gaucher-Krankheit, Duchenne/Becker-Myodystrophie, spinale Muskelatrophie, Farbenblindheit.
Schwere monogene Krankheiten können durch Pränataldiagnose und durch die Bestimmung von Mutationen bei den Eltern mit dem Atlas Genetischer Test.

Was kann ich durch einen Gentest herausfinden?
Mitochondriale Krankheiten
Verursacht durch genetische, strukturelle und biochemische Defekte in der Funktion der Mitochondrien, die zu Störungen der Gewebeatmung führen.
Wie Erbkrankheiten vererbt werden
Der menschliche Körper besteht aus Billionen von Zellen. Jede von ihnen hat einen Zellkern, der Chromosomen enthält. Jedes Chromosom besteht aus eng gewickelten Strängen von Desoxyribonukleinsäure (DNA).

Gene sind Anweisungen für den Zusammenbau von Proteinen in unserem Körper, die bestimmte Merkmale jedes Einzelnen bestimmen, wie z. B. die Augen- oder Haarfarbe.
Die meisten Körperzellen enthalten in der Regel 46 Chromosomen, die in 23 Paaren angeordnet sind. Jedes dieser 23 Paare hat ein Chromosom vom Vater und eines von der Mutter geerbt. Von den 23 Paaren sind 22 Paare bei weiblichen und männlichen Organismen identisch, und die restlichen Paare bestimmen, ob man männlich (XY) oder weiblich (XX) ist.
Mutationen, die Erbkrankheiten verursachen, können dominant oder rezessiv vererbt werden.
Dominante Vererbung bedeutet, dass nur eine Kopie des Gens – von der Mutter oder dem Vater – die Mutation (oder pathogene Genvariante) aufweisen muss, damit das Merkmal oder die Krankheit auftritt. Beim rezessiven Typ hingegen erbt eine Person zwei veränderte Kopien desselben Gens.
Autosomal-dominantes Vererbungsmuster
Beim autosomal-dominanten Erbgang ist eine Krankheit genetisch bedingt, wenn eine Person mindestens ein mutiertes Gen hat und dieses Gen nicht auf den Geschlechtschromosomen (X und Y) liegt.
Die Huntington-Krankheit und das Marfan-Syndrom sind zwei Beispiele für autosomal-dominant vererbte Krankheiten. Mutationen in den Genen BRCA1 und BRCA2, die auch mit Brustkrebs in Verbindung gebracht werden, werden nach diesem Muster vererbt.
Autosomal rezessives Vererbungsmuster
Bei der autosomal rezessiven Vererbung sind beide Kopien des Gens mutiert. Um eine autosomal rezessiv vererbte Krankheit wie zystische Fibrose, spinale Muskelatrophie oder Phenylketonurie (PKU) zu vererben, müssen beide Elternteile Träger sein.
Klassische Symptome der Marfan-Krankheit
Die klassischen Symptome der Krankheit sind eine große, schlanke Statur, eine Verkrümmung oder Skoliose der Wirbelsäule, lange Arme und Beine, die in keinem Verhältnis zum Rumpf stehen, lange dünne ‚Schwimmhäute‘ an den Fingern und unterentwickelte Muskeln. Ihre Haut ist zerbrechlich und dehnt sich leicht aus, während sie eine erhöhte Blutungsneigung haben. Die kardiovaskuläre Untersuchung zeigt eine Erweiterung des Aortenbogens und verschiedene Arten von Herzklappenfehlern.
Kurzsichtigkeit ist recht häufig und tritt bei mehr als der Hälfte aller Patienten mit Marfan-Syndrom auf, bedingt durch die sphärische Form der Linse, die veränderte Brechkraft der Hornhaut und die Verformung des Augapfels selbst.
Auch an der Iris gibt es Veränderungen, die mit einer erhöhten Gewebedehnung einhergehen. Infolgedessen bilden sich Defekte in der Iris, so genannte Kolobome, und der Winkel der vorderen Augenkammer kann durch das gedehnte Irisgewebe verschlossen werden, was zu einem Anstieg des Augeninnendrucks, d. h. zur Entwicklung eines Glaukoms führt.
Durch die Dehnung und Schwächung der Bänder, die die Linse zusammenhalten, der so genannten Cinna-Bänder, kommt es zu einer teilweisen oder vollständigen Ruptur. Entweder kommt es zu einer Subluxation der Augenlinse, bei der die Augenlinse durch einen teilweisen Riss der Bänder verschoben wird, aber noch von den verbleibenden Bändern gestützt wird. Oder die Bänder lösen sich vollständig und die Augenlinse bewegt sich nach unten in die Augenhöhle, wobei sie ihre Position frei verändern kann – die Augenlinse wird verschoben. Darüber hinaus entwickeln sich Linsentrübungen oder Katarakte früher und häufiger als bei gesunden Menschen.
Ein Glaukom entsteht, wenn der Abfluss von Augenflüssigkeit durch den Winkel der vorderen Augenkammer beeinträchtigt ist, weil eine veränderte Iris oder eine verschobene Linse die Abflusswege der Augenflüssigkeit blockiert.
Auch die Netzhaut wird überdehnt, was das Risiko einer peripheren chorioretinalen Dystrophie erhöht – einer lokalen Verdünnung der Netzhaut, die zu einer Netzhautablösung führen kann.
Korrektur und Behandlungsmethoden
Myopie kann mit einer Brille oder Kontaktlinsen korrigiert werden.
Bei grauem Star oder einer ausgeprägten Linsenverschiebung, die das Sehvermögen beeinträchtigt oder die Entwicklung eines Glaukoms begünstigt, ist ein chirurgischer Eingriff erforderlich, um die Linse zu entfernen und eine künstliche Intraokularlinse einzusetzen.
Bei Netzhautdystrophie mit hohem Risiko einer Netzhautablösung wird eine prophylaktische Laseroperation durchgeführt – kleine Laserverbrennungen werden zur Stärkung der Netzhaut in Bereichen mit Netzhautverdünnung eingesetzt. Tritt eine Ablösung auf, ist eine chirurgische Behandlung unumgänglich.
Marfan-Syndrom – Arten
Zu den häufigsten Symptomen des Marfan-Syndroms gehören:
- Skelettsystem. – Es gibt Anzeichen, die Patienten mit Marfan-Syndrom von gesunden Menschen unterscheiden. Diese Patienten neigen dazu, ungewöhnlich groß zu sein. Ihre Gliedmaßen sind deutlich verlängert und ihre Finger sind dünn und ungewöhnlich lang. Diese Form der Gliedmaßen wird als Arachnodaktylie bezeichnet. Es ist nicht ungewöhnlich, dass sich die Skoliose bereits im Kindesalter entwickelt, und der Brustkorb kann trichterförmig oder alternativ gekielt sein. Im ersten Fall kann der Brustkorb eingedrückt sein, während er im zweiten Fall vorstehen kann. An den unteren Gliedmaßen können Anomalien, wie z. B. Plattfüße, beobachtet werden.
Die Zehen können sich wie Hammerzehen verformen und abknicken. Die Gelenke sind im Allgemeinen sehr beweglich, was manchmal zu Verrenkungen führen kann. Darüber hinaus können sich all diese Anomalien als chronische Gelenk- und Muskelschmerzen äußern. Die leicht dehnbare Haut neigt dazu, Dehnungsstreifen (Striae) zu entwickeln. Manchmal kann die Stimme verändert sein, was auf einen hohen ‚gotischen‘ Gaumen, einen kleinen Kiefer oder eine Zahnfehlstellung zurückzuführen sein kann.
- Auf der visuellen Seite – Es ist nicht ungewöhnlich, dass sich die Linse aufgrund einer Schwächung des Zinnar-Bandes, an dem die Linse aufgehängt ist, verschiebt oder subluxiert (Ektopie). In diesem Fall kommt es zu verschiedenen Brechungsfehlern (Myopie, Astigmatismus). Dies äußert sich in einer Verringerung der Sehschärfe. Patienten mit Marfan-Syndrom können auch ein Glaukom und eine Netzhautablösung entwickeln.
- Herz-Kreislauf-Erkrankungen – Aufgrund der Schwächung der Bindegewebsfasern kann es zu Herzklappenerkrankungen kommen. So wird beispielsweise ein Mitral- und Aortenklappenprolaps festgestellt, der die normale Hämodynamik stört. Pathologische Veränderungen der Aortenwand führen zur Bildung von Aneurysmen oder einfach zur Vergrößerung der Aorta an der Stelle des höchsten Blutdrucks. Bei einer solchen Erkrankung besteht die Gefahr einer plötzlichen Ruptur des Aneurysmas und der Aortenwand, was durch massive innere Blutungen kompliziert wird, die oft mit dem Leben unvereinbar sind. Herz-Kreislauf-Störungen äußern sich in der Regel als Dyspnoe, Herzrhythmusstörungen, Tachykardie, Angina pectoris und erhöhte Müdigkeit. Periphere Durchblutungsstörungen können sich als anhaltendes Frösteln in den distalen Extremitäten äußern.
- Störungen der Atmung – Abnormales Bindegewebe in der Lunge kann einen Spontanpneumothorax verursachen. Luft kann aus der Lunge entweichen und sich in der Pleurahöhle ansammeln;
- Beteiligung des zentralen Nervensystems – Aufgrund eines Defekts in den Bindegewebsfasern kommt es bei Patienten mit Marfan-Syndrom zu einer Dehnung der Dura (Duralektasie). Dieser pathologische Zustand äußert sich durch Schmerzen und neurologische Anomalien im unteren Rücken und in den unteren Gliedmaßen, die sich beim Stehen verschlimmern.
Marfan-Syndrom – Diagnose in Israel
Die Diagnose des Marfan-Syndroms stützt sich auf das Vorhandensein der oben genannten Symptome. Die Diagnose wird gestellt, wenn vier verschiedene muskuloskelettale Symptome in Kombination mit Symptomen anderer Körpersysteme auftreten. Die Familienanamnese des Patienten und das Auftreten der Krankheit bei nahen Verwandten werden bei der Diagnosestellung ebenfalls in Betracht gezogen. Wenn eine Pathologie verschiedener Organe und Systeme festgestellt wird, werden die erforderlichen Untersuchungen durchgeführt: Ultraschall, CT, MRT, augenärztliche Untersuchung, Angiographie. Die Diagnose kann durch molekulargenetische Tests verifiziert werden, um das Vorhandensein des pathologischen Gens zu bestätigen.
Eine ätiologische Behandlung des Marfan-Syndroms gibt es noch nicht. Heute stehen jedoch moderne diagnostische und therapeutische Maßnahmen bereit, die Patienten mit dieser Diagnose helfen, alles zu tun, um ihr Leben zu verlängern. Für Patienten mit Marfan-Syndrom sind regelmäßige Besuche beim Kardiologen wichtig. Nach einer gründlichen Untersuchung des Patienten wird der Arzt Medikamente verschreiben, um das Risiko einer Schädigung der Herzklappen zu verringern. Dazu können Betablocker zur Senkung von Herzfrequenz, Herzschlag und Blutdruck gehören.
Bei der Behandlung des Marfan-Syndroms spielt die Vorbeugung einer Aortenruptur eine besondere Rolle. Wenn die Aneurysmaausbuchtung fortschreitet, wird ein chirurgischer Eingriff empfohlen, um das Aneurysma auszuschließen, die Wand zu verstärken oder einen Shunt anzulegen. Eine chirurgische Behandlung kann auch erforderlich sein, wenn die Wirbelsäule stark deformiert ist. Verschiedene physiotherapeutische Techniken haben eine positive Wirkung auf den Bewegungsapparat.
Eine qualitativ hochwertige symptomatische Behandlung des Marfan-Syndroms ermöglicht es den Betroffenen, ein aktives Leben zu führen.
Achtung! Alle Felder des Formulars sind Pflichtfelder. Andernfalls werden wir Ihre Informationen nicht erhalten. Alternativ können Sie auch die Adresse [email protected] verwenden.
Diagnose des Marfan-Syndroms in Israel

Patienten, die zur Behandlung kommen, werden einer umfassenden Untersuchung unterzogen. Ziel ist es, das Stadium und den Schweregrad der bereits entdeckten Pathologien zu bestimmen sowie ein Maximum an Informationen über verborgene Risiken und das Risiko weiterer Komplikationen zu erhalten. Molekulargenetische Tests werden für alle Personen empfohlen, bei denen das Marfan-Syndrom in der Familie vorkommt. Dabei wird mit 100-prozentiger Wahrscheinlichkeit ein abnormales Gen entdeckt, so dass die Behandlung so früh wie möglich beginnen kann.
Die Klinik Top Assuta verfügt über ein eigenes Diagnosezentrum, in dem alle für die Diagnose erforderlichen Tests an einem Ort durchgeführt werden können. Patienten, die auf eigene Faust kommen, werden nach dem Prinzip ‚Wer zuerst kommt, mahlt zuerst‘ untersucht, während Patienten, die ihre Reise über die offiziellen Vertreter der Klinik in ihrem Land organisiert haben, nach einem vorab genehmigten Zeitplan untersucht werden. Dieser Zeitplan sieht vor, dass alle erforderlichen diagnostischen Verfahren an drei Tagen durchgeführt werden können.
Der erste Tag
Ankunft, Treffen mit dem behandelnden Arzt, Konsultation und Untersuchung. Bei der körperlichen Untersuchung wird die Diagnose anhand von 4 Symptomen und dem Vorhandensein von Verwandten mit der gleichen Diagnose bestätigt. Insgesamt gibt es etwa 30 Anzeichen, die auf das Vorliegen einer solchen Krankheit hinweisen können. Ein erfahrener und kompetenter Arzt wird in der Lage sein, sie zu erkennen. Wenn der Arzt kein Russisch spricht, wird ein Dolmetscher zur Verfügung gestellt. Sie können den Arzt vor Ihrer Abreise auswählen, indem Sie die Lebensläufe der Fachärzte auf der Website der Klinik lesen.
Zweiter Tag
Alle Arten von Untersuchungen, Labor- und Instrumentaluntersuchungen:
Tag drei
Zur Besprechung der Untersuchungsergebnisse findet eine Konsultation statt, an der Fachärzte verschiedener Fachrichtungen teilnehmen: Kardiologe, Augenarzt, Orthopäde, Pulmologe, usw. Bei der Konsultation wird die Diagnose bestätigt und geklärt und ein Behandlungsprogramm entwickelt, das die besten Ergebnisse verspricht.
Wie hoch sind die Kosten für die Behandlung des Marfan-Syndroms in Israel?
Die Behandlung ist 30-50 % billiger als in Ländern mit einer ähnlichen Behandlungseffizienz für diese Pathologie. Es ist wichtig zu wissen, dass die Kosten für die Behandlung des Marfan-Syndroms in Israel von den individuellen Merkmalen der Krankheit, der Schädigung der verschiedenen Systeme und Organe und der Wahl der Behandlungsstrategie abhängen.
- Nachhaltige Verbesserung des Zustands des Patienten;
- Beseitigung schwerwiegender Komplikationen mit den schonendsten Mitteln, die möglich sind;
- Einsatz fortschrittlicher Techniken und innovativer Geräte;
- Erfahrene und hochqualifizierte Ärzte;
- Eine angenehme Krankenhausumgebung;
- wettbewerbsfähige Preise für die Therapie.
- Das Marfan-Syndrom auf einen Blick.
- Marfan-Syndrom bei Neugeborenen.
- Marfan-Syndrom.
- Ehlers-Danlo-Syndrom.
- Charcot-Marie-Fuß.
- Behandlung des Kurzbeinsyndroms.
- Ektrodaktylie.
- Parese des Unterkörpers.