Abnorme Veränderungen in der Hauptschlagader des Körpers, der Aorta, stellen das größte Risiko dar. Bei 65-100 % der Menschen mit Marfan-Syndrom besteht ein hohes Risiko für Läsionen im Bulbus (dem Teil der Aorta, der dem Herzen am nächsten liegt) und im aufsteigenden Bogen dieser Arterie, den Teilen, die direkt aus dem Herzen kommen. Da die innere Schicht der Gefäßwand auch Bindegewebsfasern enthält, sind diese anfällig für Abnutzung und Verschleiß, und der Blutdruck in der Aorta ist höher als in anderen Teilen des Gefäßbettes. Dies führt dazu, dass sich das Gefäß allmählich ausdehnt und es zu abnormen Blutansammlungen zwischen den Gefäßwänden kommen kann, die eine sackartige Ausbuchtung (Aneurysma) oder einen spontanen Riss der Arterie verursachen.

- Marfan-Syndrom
- Erstklassige Pflege: Was moderne Windeln leisten können
- Symptome
- Ursachen des Marfan-Syndroms
- Klassifizierung des Marfan-Syndroms
- Wie äußert sich das Marfan-Syndrom?
- Anzeichen des Marfan-Syndroms
- Diagnosemethoden für das Marfan-Syndrom
- Wie ist die Prognose für Menschen mit dieser Krankheit?
- Häufig gestellte Fragen
- Ursachen des Marfan-Syndroms
- Wie wird diagnostiziert?
- Phänotyp und Entwicklung des Marfan-Syndroms
- Behandlung des Marfan-Syndroms
- Wie äußert sich das Marfan-Syndrom?
- Behandlungsmöglichkeiten des Marfan-Syndroms
- Prognose des Marfan-Syndroms
- Leitlinien
Marfan-Syndrom
Das Marfan-Syndrom ist eine autosomal-dominante Systemerkrankung, die durch eine Unterentwicklung von Bindegewebsfasern aufgrund von Strukturdefekten im Kollagen gekennzeichnet ist. Die am häufigsten betroffenen Organe sind die Sehorgane, der Bewegungsapparat, das Herz und die Blutgefäße. Die Prognose der Krankheit hängt von der Schwere der begleitenden pathologischen Veränderungen ab. Die meisten Patienten werden nicht älter als 50 Jahre.
Die charakteristischen Symptome des Marfan-Syndroms wurden erstmals 1875 von amerikanischen Ärzten beschrieben. Benannt ist die Krankheit nach dem französischen Kinderarzt A. Marfan benannt, der die Krankheit mehrere Jahre lang erforschte.
Heute ist das Marfan-Syndrom nicht sehr häufig. Die durchschnittliche Inzidenz liegt zwischen 1 von 20 000 und 1 von 5 000 Personen. Auch dieses Syndrom ist weder rassisch determiniert noch weist es ein rassisches Muster auf. Auch dieses Problem ist weder rassisch determiniert, noch gibt es ein rassisches Muster.

Erstklassige Pflege: Was moderne Windeln leisten können
Symptome
Die Symptome des Marfan-Syndroms sind vielfältig.
Patienten, die an dieser Krankheit leiden, neigen zu einem merkwürdigen Aussehen. Sie sind groß, haben einen relativ kurzen Rumpf und übermäßig verlängerte Gliedmaßen und Oberarme. Die Arme und Beine der Patienten sind nicht nur unverhältnismäßig lang, sondern auch sehr dünn. Die Finger sind gestreckt und dünn, das Unterhautfettgewebe ist unterentwickelt und der Muskeltonus reduziert.
Der Kopf des Patienten ist lang und schmal. Häufig besteht eine Anomalie des Oberkiefers mit einer nach oben gerichteten Krümmung des harten Gaumens, was zu einem hohen Bogen führt. Die Kieferdeformität führt zu einem anormalen Biss.
Die Gelenke des Patienten sind sehr beweglich (hypermobil), und der Brustkorb ist deformiert, wobei das Brustbein nach vorne (kielförmig) oder nach innen (trichterförmig) vorsteht. Hinzu kommen verschiedene Wirbelsäulendeformitäten wie die Krümmung in der anteroposterioren Ebene.
Typisch für das Marfan-Syndrom sind verschiedene pathologische Veränderungen des Herz-Kreislauf-Systems. Diese können durch Gefäßwanddefekte wie Aneurysmen, Klappen- und Septumdefekte wie Mitralklappenprolaps, Aortenwurzelerweiterung usw. dargestellt werden. Im Jahr 2013 kündigten Forscher des Nationalen Medizinischen Forschungszentrums für kardiovaskuläre Chirurgie in Moskau, Russland, den Start eines herzchirurgischen Programms für 2013 an. Das A.N. Bakoulev Medical Centre veröffentlichte die Ergebnisse einer Studie, aus der hervorging, dass eine Aortenwurzelerweiterung bei 60 % der Patienten mit Marfan-Syndrom und ein Mitralklappenprolaps bei 91 % der Patienten auftritt.
Außerdem wirkt sich die Krankheit häufig auf die Sehorgane aus. Klinisch kann sie sich in Form von mehr oder weniger starker Myopie, Linsenektopie, Schielen, Hornhautanomalien usw. äußern.
Ursachen des Marfan-Syndroms
Die genetische Störung wird durch einen Defekt im FBN1-Gen auf dem langen Arm von Chromosom 15 verursacht. Dieses Gen kodiert das Glykoprotein Fibrillin-1, das für die Festigkeit und Elastizität des Bindegewebes verantwortlich ist. Daher sind alle Erscheinungsformen der Pathologie darauf zurückzuführen, dass die Bindegewebsstrukturen im menschlichen Körper ihre normalen Eigenschaften verlieren.
Die Mutation wird autosomal-dominant vererbt, d. h. die Kinder erhalten das pathologische Gen von Eltern, die an der Pathologie leiden. Es besteht eine 50%ige Chance, dass ein Kind die Mutation von einem Elternteil erhält (Abbildung 1). Das Syndrom wird nicht über Generationen hinweg vererbt: Gesunde Kinder von betroffenen Eltern können das Gen nicht an ihre Nachkommen weitergeben.
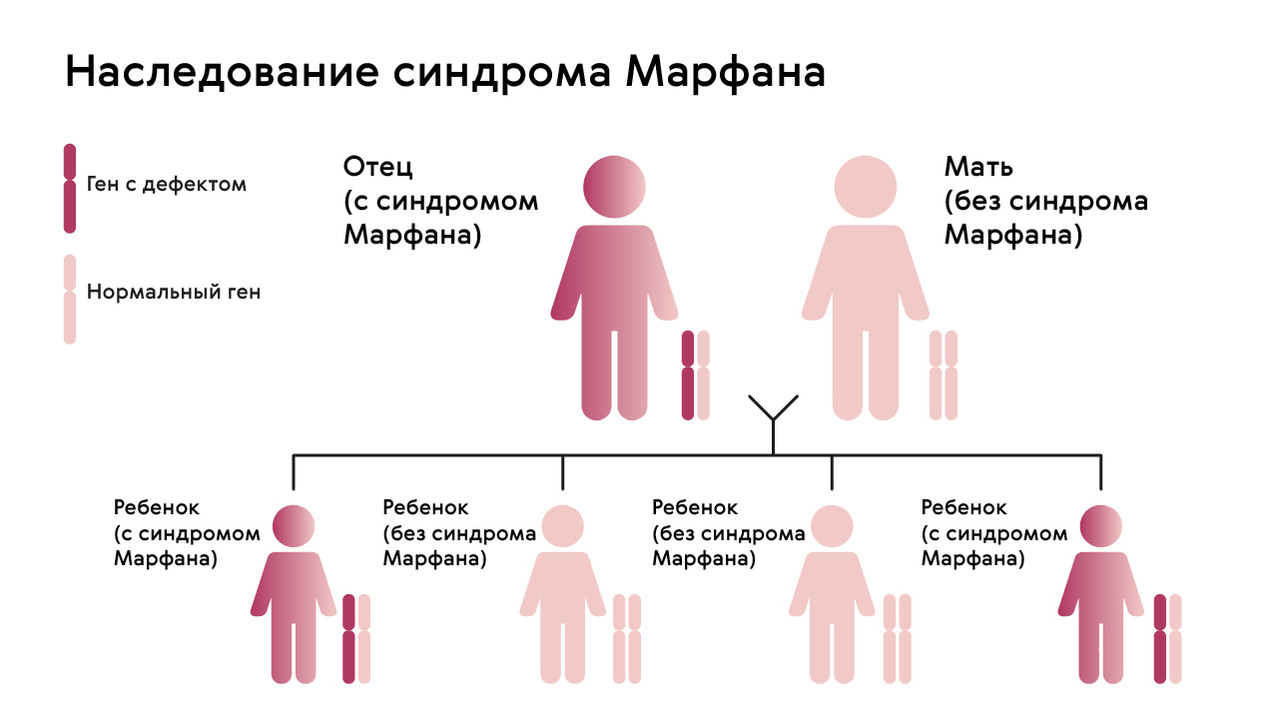
Bei etwa 25 % der Menschen mit Marfan-Syndrom ist jedoch kein Elternteil Träger der FBN1-Genanomalie: In diesem Fall entwickelt sich die Mutation spontan.
Bislang wurden keine eindeutigen Risikofaktoren für diese genetische Störung ermittelt: Die Krankheit tritt bei Männern und Frauen gleichermaßen auf, und ihre Prävalenz ist unabhängig von Rasse oder ethnischer Gruppe. Die Prävalenz dieser Pathologie liegt bei etwa 1 zu 5-10 000.
Wenn die klinischen Manifestationen der Mutation eindeutig sind, kann die Krankheit bereits in den ersten Lebensmonaten vermutet werden. Milde Formen der Krankheit treten jedoch häufig erst im Erwachsenenalter auf, wenn der Patient die Ärzte wegen der verschiedenen Erscheinungsformen des Syndroms konsultiert.
Wichtig!!! Buchen Sie einen Termin für einen Gentest nicht als medizinische Untersuchung. Die Suche nach einem ‚Defekt‘ im FBN1-Gen ist nur dann gerechtfertigt, wenn die Krankheit charakteristische Symptome hervorruft: Eine asymptomatische Vererbung dieser Mutation ist nicht möglich. Wenn ein Elternteil diese Diagnose hat, sollte die werdende Mutter vor der Geburt einen Gentest durchführen lassen. Auf diese Weise lässt sich im Voraus feststellen, ob die Anomalie auf das Kind übertragen wurde.
Klassifizierung des Marfan-Syndroms
Je nach den klinischen Erscheinungsformen der genetischen Mutation gibt es verschiedene Formen der Krankheit.
Es gibt zwei klinische Hauptformen der Pathologie:
- Mild. Diese Patienten haben ‚Glück‘, weil die Anomalie nur ein oder zwei Körpersysteme betrifft und ihre Symptome mild sind. Die Betroffenen können trotz der Krankheit ein nahezu normales Leben führen.
- Schwerwiegend. In diesen Fällen sind drei oder mehr Körpersysteme betroffen oder ein System ist erheblich beeinträchtigt.
Je nach Ausprägungsgrad gibt es leichte, mittlere und schwere Formen des Marfan-Syndroms. Schwere Pathologien sind mit einer Inzidenz von etwa 1 von 25-50.000 Menschen deutlich seltener.
Die Art des Krankheitsverlaufs spielt eine entscheidende Rolle bei der Bestimmung der Prognose der Krankheit:
- Progredient. In diesem Fall treten ständig neue Krankheitssymptome auf, der Schweregrad der Erkrankung nimmt zu und das Risiko tödlicher Komplikationen steigt mit jedem Lebensjahr des Patienten.
- Stabil. Dieses Krankheitsbild gilt als das günstigste: Bei Patienten mit stabilen Symptomen des Marfan-Syndroms bleibt das klinische Bild während des gesamten Lebens weitgehend unverändert.
Es werden drei verschiedene, aber ähnliche Krankheiten unterschieden:
- Marfan-Syndrom – eine milde Form der Krankheit mit einem positiven Gentest.
- Marfan-Syndrom – das klassische Krankheitsbild mit gesicherter familiärer Vererbung.
- Marfan-Syndrom – eine Manifestation der Bindegewebspathologie ohne genetische Mutation.
Die ersten Symptome der Krankheit treten in der Regel in der Kindheit auf. Wenn der Patient das Jugendalter erreicht, wird das Fortschreiten der Krankheit aufgrund der Mutation des FBN1-Gens deutlich.
Wie äußert sich das Marfan-Syndrom?
Bei der milderen Form sind 1-2 Körpersysteme betroffen (z. B. Muskel-Skelett- und/oder Sehorgane), und die Symptome sind mild. Trotz der Anomalien führen diese Patienten ein nahezu normales Leben. Bei der schweren Form sind 3 oder mehr Körpersysteme betroffen, oder es kommt zu erheblichen Veränderungen in einem der Systeme.
Ein und derselbe Gendefekt äußert sich auf unterschiedliche Weise, von leichten Veränderungen bis hin zu schweren Organstörungen.
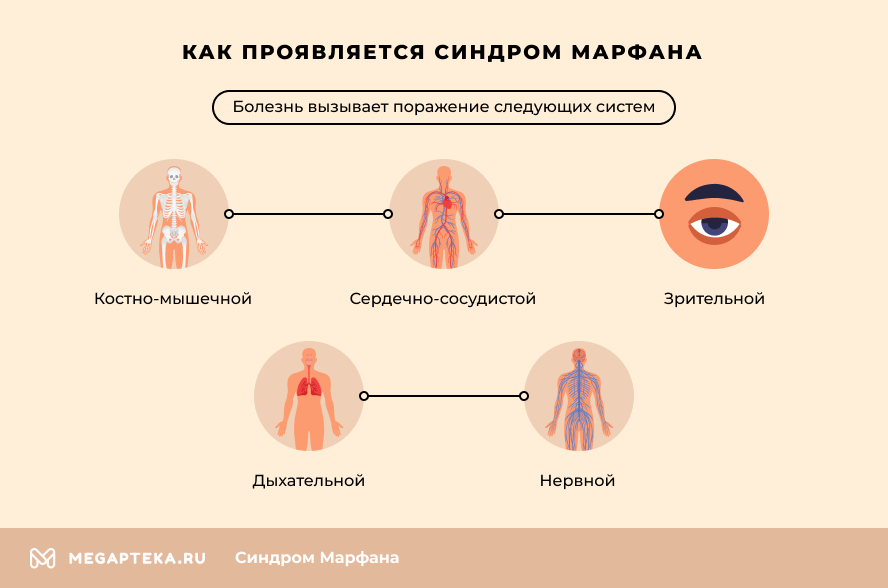
- Muskuloskelettales System – die Patienten zeichnen sich durch Hochwuchs, abnorme Dünne und ein längliches Gesicht aus. Weitere auffällige Symptome sind verlängerte und dünne Finger und eine unregelmäßige Armlänge. Die Körperhaltung ist in den meisten Fällen abnormal, mit Skoliose oder Kyphose. Häufig haben die Patienten Plattfüße und eine erhöhte Gelenkbeweglichkeit.
- Herz-Kreislauf-System (CVS) – das Syndrom verursacht Herzrhythmusstörungen, Mitralklappeninsuffizienz und verschiedene Herzfehler. Besonders gefährlich sind abnorme Veränderungen der Aorta. Bei den meisten Patienten besteht ein erhöhtes Risiko einer Dilatation des aufsteigenden Teils und des Klappenrings sowie der Bildung eines Aneurysmas.
- Sehkraft – schwere Myopie, Linsensubluxation oder Veränderungen der Linsenposition sind am häufigsten. Es besteht auch ein erhöhtes Risiko für Netzhautablösungen. Glaukom und Katarakt entwickeln sich oft schon in jungen Jahren.
- Atmungsorgane – eine abnorme Hypertrophie des Bindegewebes in der Lunge führt zu Bronchokonstriktion und Lungenfibrose. Häufig führt eine genetische Mutation zur Entwicklung von Asthma bronchiale.
- Nervensystem – in den meisten Fällen gibt es keine Anomalien in den Gehirnstrukturen. Eine Vergrößerung der Bindegewebskapsel um das Rückenmark führt jedoch zu Störungen der Beinbewegungen, der Blasen- und Darmfunktion. Eine hohe Adrenalinausschüttung führt häufig zu erhöhter nervlicher Erregbarkeit und Hyperaktivität.
Nierenvorfall, Gebärmuttervorfall, Krampfadern und gastrointestinale (GI) Anomalien können ebenfalls beim Marfan-Syndrom auftreten.
Anzeichen des Marfan-Syndroms
- Familienanamnese – eine Erklärung für die Erkrankung der Eltern;
- Körperliche Diagnose – Vorhandensein typischer Symptome, einschließlich der Merkmale der anthropometrischen Indizes;
- Untersuchungsdaten – EKG (Elektrokardiographie), EchoCG (Echokardiographie) des Herzens, MRT (Magnetresonanztomographie) des Gehirns und der Wirbelsäule, Röntgenaufnahmen, ophthalmologische Untersuchung;
- Gentests.
Diagnosemethoden für das Marfan-Syndrom
Die Diagnose des Marfan-Syndroms stützt sich auf klinische Befunde, Gentests und Echokardiographie oder MRT. Mit der Echokardiographie und der MRT werden abnorme Veränderungen der Aortenwurzel und der Klappen festgestellt. Abnormale Veränderungen der Linse lassen sich mit einer Spaltlampe erkennen, und eine Vergrößerung des Duralsacks im Rückenmark lässt sich mittels MRT nachweisen. Bei der Untersuchung des Auges sieht der Augenarzt das typische Zeichen für eine genetische Anomalie in Form einer Linsensubluxation.
In manchen Fällen ist es jedoch schwierig, eine korrekte Diagnose zu stellen, da viele Patienten nicht die auffälligen Symptome des Marfan-Syndroms aufweisen. Es kann auch sein, dass sie keine Veränderungen in der Biochemie oder Histologie aufweisen. In solchen Fällen sollten sich die Ärzte auf die Anamneseerhebung, die objektive Untersuchung und genetische Tests konzentrieren.
Wie ist die Prognose für Menschen mit dieser Krankheit?
Dank der Fortschritte in der modernen Medizin ist die Sterblichkeitsrate dieser Patienten heute deutlich geringer. Während sie früher im Durchschnitt nicht länger als 48 Jahre lebten, ist ihre Lebenserwartung heute fast genauso hoch wie die von ’normalen‘ Menschen. Natürlich sind kardiale und vaskuläre Komplikationen immer noch ein Problem.
- Bei der Verschreibung von Betablockern;
- die chirurgische Korrektur von Klappen- und Aortenanomalien;
- chirurgische Korrektur von Anomalien der Wirbelsäule.
Von den Betablockern ist es ratsam, den Patienten Propranolol oder Atenolol zu verschreiben. Dadurch werden schwerwiegende vaskuläre und kardiale Komplikationen verhindert. Betablocker reduzieren die Intensität der Herzmuskelkontraktion, wodurch die Belastung des Herzmuskels verringert, der Prozess der Aortendissektion gestoppt und das Risiko der Entwicklung eines Aneurysmas verringert wird. Wenn das Aneurysma eine kritische Größe erreicht, ist eine Operation angezeigt.
Die Skoliose wird in der Regel konservativ mit einer Wirbelsäulenfixierung behandelt, doch wenn die Wirbelsäule um 40 Grad oder mehr gekrümmt ist, wird eine Operation bevorzugt.
Alle Patienten mit Marfan-Syndrom sollten jährlich von einem Neurologen, einem Kardiologen und einem Augenarzt untersucht werden, ggf. mit genetischer Beratung.
Häufig gestellte Fragen
Kann ich verhindern, dass mein Kind ein Marfan-Syndrom bekommt?
Wenn bei mindestens einem Ehepartner das Marfan-Syndrom in der Familie vorkommt, sollte sich das Paar vor der Empfängnis einem Gentest unterziehen. Nach der Schwangerschaft wird ein pränatales Screening durchgeführt: Ultraschall des Fötus und eine Reihe biochemischer Tests: mütterliches Serum, Fruchtwasser, Chorionbiopsie, Anzahl der Zellen in der Plazenta und Nabelschnurblut.
Nein. Die Diagnose eines Marfan-Syndroms bei einem Wehrpflichtigen ist ein Grund für die Entlassung aus dem Militärdienst.
Welche Präventivmaßnahmen gibt es beim Marfan-Syndrom?
Die Patienten werden ein Leben lang von einem Arzt überwacht. Vorbeugende Maßnahmen zielen darauf ab, Komplikationen zu vermeiden. Zu diesem Zweck werden herzchirurgische Eingriffe vorgenommen, eine Pharmakotherapie zur Beseitigung des Risikos einer Gefäßthrombose durchgeführt, eine Antibiotikatherapie durchgeführt usw.
Ursachen des Marfan-Syndroms
Die Krankheit tritt bei 1 von 10 000 Menschen auf. Der Entstehung des Marfan-Syndroms liegt ein Defekt in einem Gen auf Chromosom 15 zugrunde, das die Struktur von Fibrillin bestimmt. Dieses Protein ist der Hauptbestandteil der Elastin-assoziierten Mikrofibrillen. Letztere finden sich in den meisten Bindegeweben, großen Blutgefäßen und Bändern, die die Linse stützen. Die Krankheit wird autosomal-dominant vererbt: Wenn ein Elternteil die Krankheit hat, besteht eine 50 %ige Chance, dass das Kind die Pathologie entwickelt. Es gibt Fälle, in denen ein gesundes Paar ein Kind mit dieser genetischen Anomalie hat: eine spontane Mutation tritt in 1 von 4 Fällen auf.
- kielförmige oder trichterförmige Krümmung des Brustkorbs, Wirbelsäulendeformität
- überproportionaler Rumpf, hohe Statur
- lange (gekrümmte) Finger (wenn das Handgelenk positiv ist, überlappen sich Daumen und Zeigefinger, wenn die andere Hand gefesselt ist)
- Vergrößerung des Bewegungsumfangs der Gliedmaßen
- abnorme Gelenkbeweglichkeit
- Plattfuß-Deformität
- Gewichtsabnahme
- gewölbter Gaumen, Zahndefekte
- abnormes Gesicht (Dolichocephalie, Unterentwicklung des Jochbeins, eingefallene Augen)
- Sehstörungen aufgrund einer Ektopie (Verlagerung) der Linse, einer abnormen Abflachung der Hornhaut, Kurzsichtigkeit usw.
- Kurzatmigkeit, Schwäche
- Hautstriae
- Wiederkehrende Leistenbrüche, usw.
Wie wird diagnostiziert?
Der Diagnosealgorithmus hängt von den beteiligten Systemen ab. Es sind Konsultationen mit folgenden Spezialisten erforderlich: Genetiker, Augenarzt, Kardiologe, Pneumologe, Gastroenterologe.
Die instrumentelle Untersuchung stützt sich auf:
- Röntgenaufnahmen oder Computerscans von Händen, Füßen, Brustkorb, Wirbelsäule, Hüftknochen und Schädel.
- Magnetresonanztomographie (MRT) – wenn wir Veränderungen an inneren Organen diagnostizieren wollen: Anomalien der Eingeweide, Herzfehler, Nieren usw. Die Magnetresonanzangiographie des Brustkorbs ermöglicht eine vollständige Beurteilung des Arteriensystems.
- EKG, Ultraschall, transthorakale Echokardiographie (EchoCG) von Herz und Gefäßen;
Phänotyp und Entwicklung des Marfan-Syndroms
Marfan-Syndrom – ist eine Multisystemerkrankung mit Skelett-, Augen-, Herz-Kreislauf-, Lungen-, Haut- und anderen Anomalien. Zu den Skelettanomalien gehören sehr hohe Werte (Verhältnis Armspanne zu Körpergröße >1,05; Verhältnis oberes zu unteres Segment
Anomalien Augen umfassen Linsensubluxation, Hornhautabflachung, Verlängerung des Augapfels und Irishypoplasie. Zu den kardiovaskulären Anomalien gehören Mitralklappenprolaps, Aortenregurgitation sowie Dilatation und Dissektion des Aneurysmas der aufsteigenden Aorta. Zu den pulmonalen Anomalien gehören Spontanpneumothorax und Alveolarerweiterung. Zu den kutanen Anomalien gehören atrophische Furchen und wiederkehrende Hernien. Zu den duralen Anomalien gehören Ausbuchtungen der Dura in der Sakralregion.
Die meisten Anzeichen des Marfan-Syndroms treten mit zunehmendem Alter auf. Skelettanomalien, wie Sternumanomalien und Skoliose, verschlimmern sich mit dem Wachstum der Knochen. Eine Linsensubluxation tritt häufig in der frühen Kindheit auf, kann sich aber auch in der Adoleszenz entwickeln.
Mit erhöhter Häufigkeit bei Marfan-Syndrom Netzhautablösung, Glaukom und Katarakt sind häufig. Herz-Kreislauf-Komplikationen treten in jedem Alter auf und entwickeln sich im Laufe des Lebens.
Die Hauptursachen für einen vorzeitigen Tod sind. Tod von Patienten mit Marfan-Syndrom sind Herzversagen aufgrund von Klappeninsuffizienz sowie Aortenaneurysma und -ruptur. Durch Verbesserungen in der chirurgischen und therapeutischen Versorgung von Aortenaneurysmen hat sich die Überlebensrate jedoch verbessert. Von 1972 bis 1993 stieg das erwartete Überlebensalter für 50 % der Patienten von 49 auf 74 Jahre bei Frauen und von 41 auf 70 Jahre bei Männern.
Auffälligkeiten Phänotypische Erscheinungsformen des Marfan-Syndroms:
– Alter des Auftretens: frühe Kindheit
– Überproportional hoch
– Skelettale Anomalien
– Ektopie (Subluxation) der Linse
– Mitralklappenprolaps
– Aortenaneurysma und Ruptur
– Spontan-Pneumothorax
– Hernie der lumbosakralen Scheide
Behandlung des Marfan-Syndroms
Das Marfan-Syndrom – ist eine klinische Diagnose, die durch das Vorhandensein von spezifischen Symptomen gestellt wird. Die Bestätigung des Marfan-Syndroms durch den Nachweis von Mutationen im FBN1-Gen ist derzeit nicht praktikabel, da die extreme Heterogenität der Allele die Identifizierung der ursächlichen Mutation in jeder Familie extrem zeitaufwändig macht und die Korrelation zwischen Genotyp und Phänotyp nicht zuverlässig ist. Die Mutationsanalyse hat eine geringe Sensitivität und Spezifität für das Marfan-Syndrom, was ihren klinischen Nutzen einschränkt.
Für Marfan-Syndrom gibt es keine wirksame Behandlung; daher konzentriert sich die Behandlung auf die Prävention von Komplikationen und die symptomatische Behandlung. Die augenärztliche Versorgung umfasst regelmäßige Kontrolluntersuchungen, die Korrektur von Kurzsichtigkeit und häufig den Austausch von Linsen. Die orthopädische Versorgung besteht aus einer verstärkenden Behandlung oder einer chirurgischen Korrektur der Skoliose. Die Behandlung von Anomalien des Brustbeins ist hauptsächlich kosmetisch.
Physiotherapie Kann die Instabilität der Gelenke ausgleichen. Herz-Kreislauf-Probleme werden durch eine Kombination aus therapeutischen und chirurgischen Maßnahmen behandelt. Therapeutische Maßnahmen zielen darauf ab, eine Aortenwurzeldilatation zu verhindern oder zu verzögern, indem die Herzparameter gesenkt werden, der Blutdruck und die ventrikuläre Auswurfkraft mit Betablockern gesenkt werden und die Teilnahme an Kontaktsportarten, Leistungssport und isometrischen Übungen eingeschränkt wird.
Prophylaktischer Ersatz der Wurzel Eine Behandlung der Aortenregurgitation ist angezeigt, wenn die Aortendilatation oder Aortenregurgitation schwerwiegend genug ist. Die meisten Patienten werden heute mit einem supraklavikulären Aortenwurzelersatz behandelt, der keine kontinuierliche Verabreichung von Antikoagulantien erfordert.
Hämodynamik Schwangerschaftsbedingte Veränderungen können zu einer fortschreitenden Aortendilatation oder Dissektion führen. Man geht davon aus, dass die Aortendissektion auf hormonelle Veränderungen, das erhöhte Blutvolumen und die erhöhte Herzleistung im Zusammenhang mit Schwangerschaft und Geburt zurückzuführen ist. Aktuelle Forschungsergebnisse deuten darauf hin, dass das Risiko einer Schwangerschaft zu hoch ist, wenn die Breite der Aortenwurzel mehr als 4 cm beträgt. Frauen können sich vor einer Schwangerschaft für einen supraklavikulären Aortenersatz entscheiden.
Wie äußert sich das Marfan-Syndrom?
Zunächst einmal fällt das besondere Aussehen der Patienten auf. Sie sind groß, haben lange Arme und Beine, die in keinem Verhältnis zu anderen Körperteilen stehen, sowie dünne und längliche Finger. Das Unterhautfettgewebe ist unterentwickelt und der Muskeltonus ist reduziert. Das Gesichtsskelett ist lang und schmal. Charakteristisch sind auch ein hochgewölbter Gaumen und verschiedene Okklusionsanomalien.
Häufig zeigen Patienten mit Marfan-Syndrom eine übermäßige Beweglichkeit der Gelenke und verschiedene strukturelle Veränderungen der Wirbelsäule und des Brustkorbs.
Es kann auch zu Anomalien des Herzens und der Blutgefäße kommen. Die Krankheit kann eine Aortendilatation, Aneurysmen und verschiedene angeborene Herzfehler wie einen Ventrikelseptumdefekt aufweisen. Die Krankheit geht häufig mit Herzrhythmusstörungen einher.
Typischerweise tritt das Marfan-Syndrom mit verschiedenen Augenanomalien in Erscheinung. Im Jahr 2012 führten Forscher des pädiatrischen medizinischen Instituts in Taschkent eine Studie durch, aus der hervorging, dass bei 50-80 % der Patienten Sehstörungen auftreten, oft als eines der ersten Symptome der Krankheit.
Zu den klinischen und morphologischen Augenanomalien beim Marfan-Syndrom können Myopie, ektopische Linse, Unterentwicklung der Iris, Schielen usw. gehören.
Darüber hinaus können auch andere innere Organe wie das zentrale Nervensystem, das bronchopulmonale System, die Haut usw. betroffen sein.
Behandlungsmöglichkeiten des Marfan-Syndroms

Aufgrund des angeborenen Charakters des Marfan-Syndroms gibt es keine Möglichkeit einer vollständigen Heilung. Die gesamte Behandlung ist symptomatisch und konzentriert sich weitgehend darauf, das Fortschreiten der Krankheit zu stoppen.
Zur Verbesserung der kardiovaskulären Funktion können Betablocker, Angiotensin-Converting-Enzyme-Hemmer usw. verschrieben werden. Eine chirurgische Behandlung, z. B. eine rekonstruktive Aortenoperation oder ein Klappenersatz, kann durchgeführt werden, wenn strukturelle Veränderungen am Herzen oder an den Gefäßen erkennbar sind.
Die daraus resultierenden Augenerkrankungen können mit Brillen oder Kontaktlinsen und Operationen zur Beseitigung von Glaukom, Katarakt usw. korrigiert werden.
Schwerwiegende Veränderungen des Bewegungsapparats können ebenfalls chirurgische Eingriffe wie Brustwandrekonstruktionen, Operationen zur Stabilisierung der Wirbelsäule usw. erforderlich machen.
Prognose des Marfan-Syndroms
Die Fortschritte in der Therapie und die regelmäßige Überwachung haben die Lebensqualität verbessert und die Sterblichkeit verringert. Die Lebenserwartung ist von 48 Jahren im Jahr 1972 auf eine annähernd normale Lebenserwartung derjenigen gestiegen, die eine angemessene medizinische Versorgung erhalten. Die durchschnittliche Lebenserwartung der Patienten ist jedoch immer noch niedriger, was vor allem auf kardiale und vaskuläre Komplikationen zurückzuführen ist. Diese geringere Lebenserwartung kann für Jugendliche und Familien eine emotionale Belastung darstellen.
Die Behandlung des Marfan-Syndroms konzentriert sich auf die Prävention und das Management von Komplikationen.
Bei sehr großen Mädchen kann die Induktion einer frühzeitigen Pubertät im Alter von 10 Jahren mit Östrogenen und Progesteron das Wachstum im Erwachsenenalter einschränken.
Alle Patienten sollten regelmäßig Betablocker (z. B. Atenolol, Propranolol) erhalten, um kardiovaskuläre Komplikationen zu vermeiden. Diese Medikamente verringern die Kontraktilität des Herzmuskels und den Pulsdruck und reduzieren das Fortschreiten des Aortenwurzelaneurysmas und das Risiko einer Aortendissektion. Auch Angiotensin-II-Rezeptorblocker können eingesetzt werden.
Bei einem Aortendurchmesser von > 5 cm (bei Kindern kleiner) wird eine prophylaktische Operation empfohlen. Schwangere Frauen haben ein besonderes Risiko für Aortenkomplikationen; eine empfohlene Aortenrekonstruktion sollte vor der Empfängnis besprochen werden. Schwere Klappeninsuffizienzen werden ebenfalls operativ rekonstruiert. Prophylaxe der bakteriellen Endokarditis Die infektiöse Endokarditis (IE) ist eine Infektion des Endokards, die in der Regel bakteriell (häufiger durch Streptokokken oder Staphylokokken) oder durch Pilze verursacht wird. Sie kann sich durch Fieber, Herzgeräusche und Petechien äußern. Weiter lesen Nicht angezeigt vor invasiven Eingriffen, außer bei Patienten mit künstlichen Herzklappen oder infektiöser Endokarditis (Eingriffe, die eine prophylaktische Antibiotikatherapie bei Patienten mit hohem Endokarditisrisiko erfordern, US Empfohlene Endokarditisprophylaxe bei zahnärztlichen Eingriffen und Eingriffen der oberen Atemwege* Empfohlene Endokarditisprophylaxe bei zahnärztlichen Eingriffen und Eingriffen der oberen Atemwege ).
Leitlinien
Das Marfan-Syndrom wird durch eine autosomal-dominante Mutation des Gens verursacht, das für das Glykoprotein Fibrillin-1 kodiert, das ein Hauptbestandteil der Mikrofibrillen ist, so dass mehrere Deformierungen und Defekte möglich sind.
Die Manifestationen sind sehr unterschiedlich, aber die wichtigsten strukturellen Defekte betreffen das kardiovaskuläre und das muskuloskelettale System sowie das visuelle System, was zu der typischen Kombination aus verlängerten Gliedmaßen, Aortenwurzeldilatation und Linsenverschiebung führt.
Um strukturelle Anomalien zu erkennen, sollte eine diagnostische Bildgebung des Skelett-, Herz-Kreislauf- und Augensystems durchgeführt werden.
Alle Patienten sollten mit Betablockern behandelt werden, um Komplikationen an der Aorta zu verhindern; andere Komplikationen sollten behandelt werden, sobald sie auftreten.
Lesen Sie mehr:- Das Marfan-Syndrom auf einen Blick.
- Marfan-Syndrom bei Neugeborenen.
- Marfan-Syndrom – Zusammenfassung.
- Ehlers-Danlo-Syndrom.
- Behandlung des Kurzbeinsyndroms.
- Syndrom des Nervus tibialis.
- Fußwurzel- und Mittelfußgelenke.
- Wie man einen Klumpfuß bei einem Kind korrigiert.