Medications used to treat symptoms suggestive of cardiovascular disease include beta-blockers, angiotensin-converting enzyme inhibitors, and calcium channel blockers. Valvular insufficiency, a thoracic aortic aneurysm five centimeters or more in diameter, and aortic dissection are indications for surgical intervention.

- Marfan syndrome
- Care at the highest level: what modern diapers can do
- symptoms
- Causes of Marfan Syndrome
- How is it diagnosed?
- Causes of Marfan Syndrome
- Classification of Marfan syndrome
- Symptoms of Marfan Syndrome
- Musculoskeletal system
- Why should a doctor be consulted when detecting symptoms of Marfan syndrome?
- Marfan Syndrome – a disease of geniuses?
- Treatment of Marfan syndrome
- What Should Doctors Know About Marfan Syndrome?
- Symptoms from different organs and systems
- Main signs of pathology
- How to accurately diagnose a disease in a child?
- Diagnosis of Marfan Syndrome
- Prognosis in Marfan syndrome
- What are the symptoms of Marfan Syndrome?
- How is Marfan syndrome treated?
- How does Marfan syndrome manifest itself?
- Treatment principles for Marfan syndrome
Marfan syndrome
Marfan syndrome is a systemic autosomal dominant disorder characterized by underdevelopment of connective tissue fibers due to structural defects in collagen. The most commonly affected organs are the eyes, the musculoskeletal system, the heart and the blood vessels. The prognosis of the disease depends on the severity of the accompanying pathological changes. Most patients do not live longer than 50 years.
The characteristic symptoms of Marfan syndrome were first described by American doctors in 1875. The disease is named after the French pediatrician A. Marfan, who researched the disease for several years.
Today, Marfan syndrome is not very common. The average incidence is between 1:20,000 and 1:5,000, with men and women being affected equally. There is also no racial pattern.

Care at the highest level: what modern diapers can do
symptoms
The symptoms of Marfan syndrome are varied.
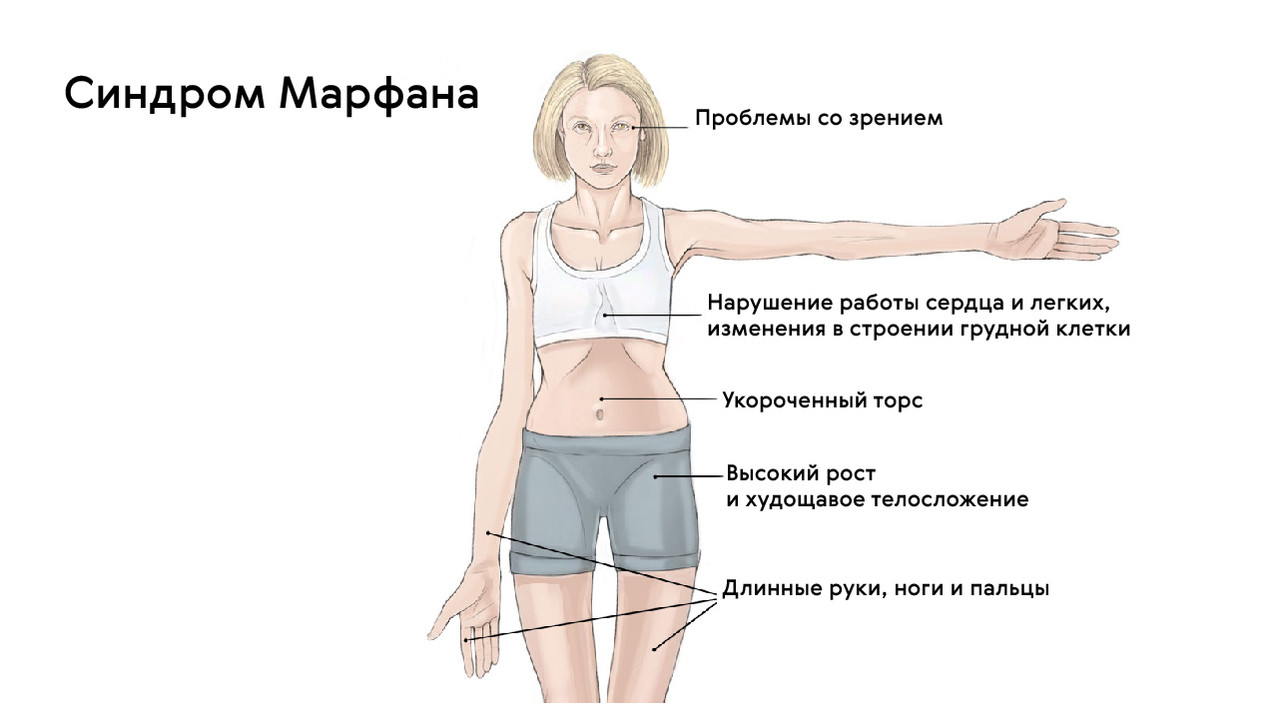
Patients with this disease have a striking appearance. They are large, with a relatively short trunk and overly elongated upper and lower limbs. Patients' arms and legs are not only disproportionately long, but also very thin. The fingers are stretched and thin, the subcutaneous fat tissue is underdeveloped, and muscle tone is reduced.
The patient's head is long and narrow. It is not uncommon for there to be an anomaly in the upper jaw, in which the hard palate bulges upwards and forms a high arch. The irregular bite is the result of misaligned jaws.
The patient's joints are highly mobile (hypermobile) and the chest is deformed, with the breastbone protruding forward (keel shape) or falling inward (funnel shape). There are also various spinal deformities such as curvature in the anteroposterior plane.
Marfan syndrome is characterized by various anomalies in the cardiovascular system. These can manifest themselves in the form of vascular wall defects such as aneurysms, valve and septum defects such as mitral valve prolapse, aortic root enlargement and so on. In 2013, researchers from the National Medical Research Center for Cardiovascular Surgery AN Bakulev published a number of studies on the development of cardiac aneurysms. The Bakoulev National Medical Research Center published a study showing that 60 % of the patients with Marfan syndrome have aortic root dilatation and 91 % have mitral valve prolapse.
In addition, visual disturbances are a common consequence of the disease. Clinically, this can manifest itself as myopia of varying degrees of severity, lens ectopia, strabismus, corneal abnormalities, etc.
Causes of Marfan Syndrome
It occurs in 1 in 10,000 people. The development of Marfan syndrome is based on a genetic defect on chromosome 15, which determines the structure of fibrillin. This protein is the main component of elastin-associated microfibrils. The latter are found in most connective tissues, major blood vessels, and ligaments that support the lens. The disease is inherited in an autosomal dominant manner: if one parent has the disease, there is a 50% chance that the child will develop the pathology. There are cases when a healthy couple gives birth to a child with this genetic anomaly: spontaneous mutation occurs in 1 out of 4 cases.
- keel or funnel curvature of the chest, spinal deformity
- disproportionate torso, tall stature
- long (curved) fingers (when the wrist is positive, the thumb and forefinger overlap when the other hand is tied)
- Increasing the range of motion of the limbs
- abnormal joint mobility
- flat valgus deformity of the foot
- lack of weight
- arched palate, deformity of the dentition
- atypical face (dolichocephaly, underdevelopment of cheekbones, sunken eyes)
- Vision problems caused by an ectopic lens, abnormal corneal flattening, myopia, etc.
- Dyspnea, weakness
- skin striae
- Recurring hernias, etc.
How is it diagnosed?
The diagnostic treatment algorithm depends on the affected systems. Consultation of the following specialists is required: geneticist, ophthalmologist, cardiologist, pulmonologist, gastroenterologist.
The instrumental study is based on:
- X-rays or computer images of hands, feet, chest, spine, hip bones, and skull.
- Magnetic resonance imaging (MRI) – when we want to diagnose changes in internal organs: abnormalities of the intestine, heart defects, kidneys, etc. Thoracoabdominal magnetic resonance angiography allows a complete assessment of the arterial system.
- ECG, ultrasound, transthoracic echocardiography (echocardiography) of the heart and vessels;
Causes of Marfan Syndrome
Marfan syndrome is an autosomal dominant congenital anomaly with marked pleiotropism, variable expression, and high penetrance. Underlying Marfan syndrome are mutations in the FBN1 gene, which is responsible for the synthesis of fibrils, an essential structural protein of the intercellular matrix that confers elasticity and contractility on connective tissue. The anomaly and lack of fibrillin in Marfan syndrome leads to impaired formation of fibrous structures, loss of strength and elasticity of connective tissue and inability to withstand physiological loads. The elastic vessel walls and ligaments (particularly the aorta and cinnamon ligament of the eye, which contain the highest proportion of fibrillin) are more susceptible to histological changes.
The wide phenotypic spectrum of Marfan syndrome (from mild, difficult to distinguish to severe, rapidly progressive forms) is due to the diversity of mutations in the FBN1 gene (more than 1,000 types) as well as the presence of mutations in other genes ( e.g., the transforming growth factor gene TGFBR-2). Genetic testing shows a familial mode of inheritance in 75 percent of Marfan syndrome cases and a primary mutation in the remaining cases. The risk of having a child with Marfan syndrome increases with the age of the father (especially after the age of 35).
Classification of Marfan syndrome
There are different forms of Marfan syndrome depending on the number of systems affected
- Sterile – with slight changes in 1 or 2 systems
- difficult – with slight lesions in 3 systems; severe lesions in at least 1 system; severe lesions in 2-3 or more systems.
The severity of changes in Marfan syndrome can be mild, moderate, or severe. A distinction is made between progressive and stable Marfan syndrome.
Symptoms of Marfan Syndrome
The symptoms of the genetic defect can range from minor changes in the connective tissue to severe disturbances of essential bodily functions. In other words, the external manifestations of the anomaly can vary greatly from patient to patient, despite the same genetic defect.
The classic triad of Marfan syndrome consists of skeletal abnormalities, lens displacement, and aortic dissection (Figure 2). In addition, systemic connective tissue damage in patients causes abnormalities in almost every organ and system in the body.
Musculoskeletal system
The severity of the musculoskeletal symptoms depends on the severity of the case and the patient's physical characteristics.
People with Marfan syndrome are exceptionally tall: the children tend to tower over all family members. Abnormal arm length is a common finding, particularly in children who have arms that are longer than their body.
A noticeable symptom is abnormally elongated and thin fingers, so-called spider fingers (arachnodactyly) (Figure 3).
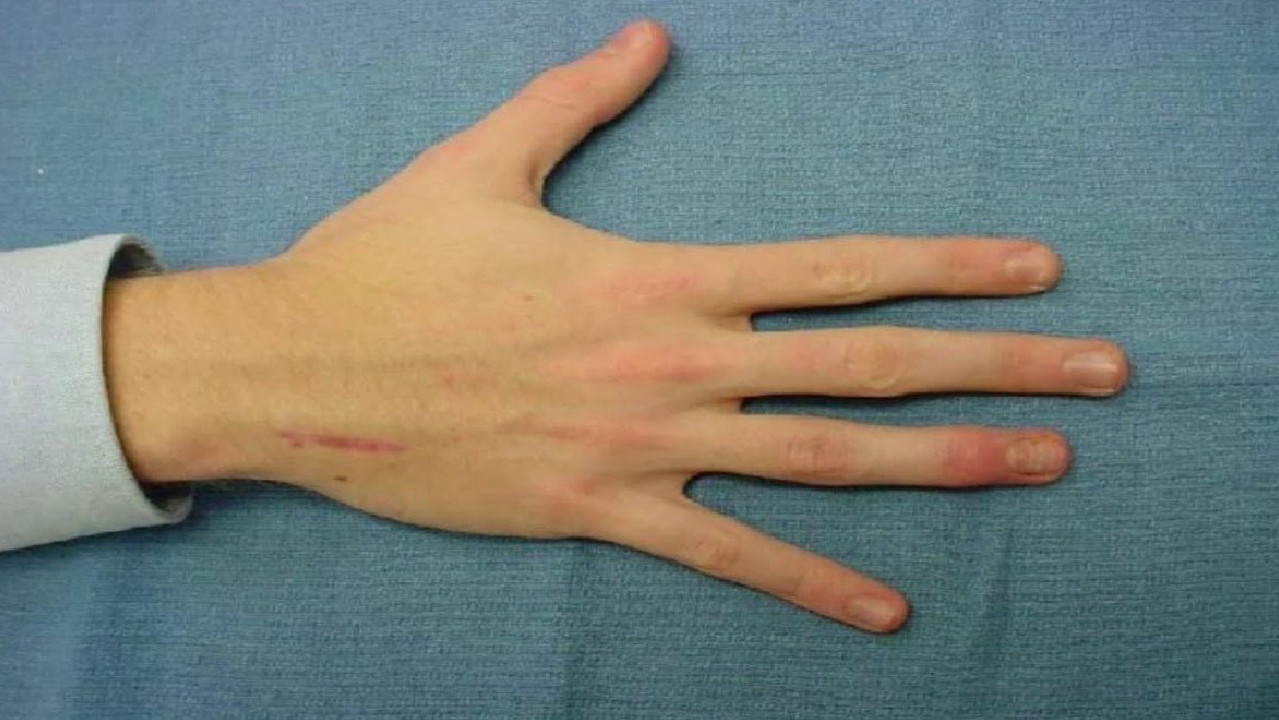
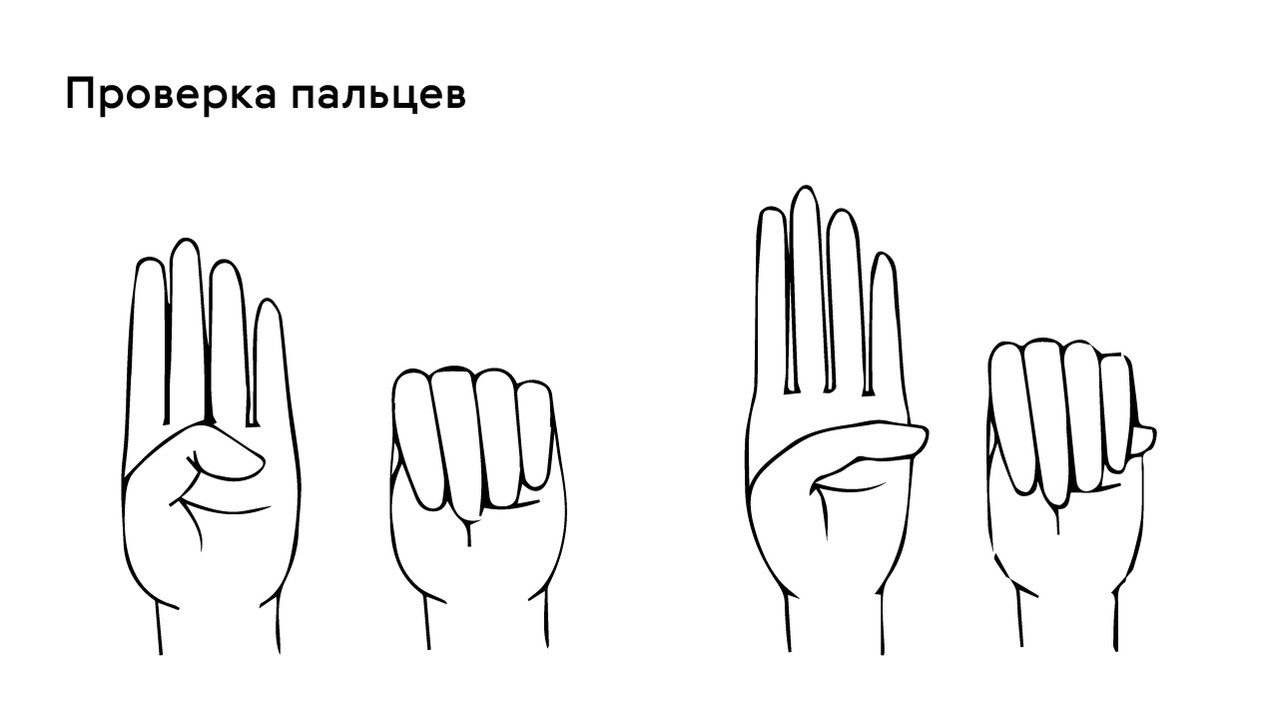
The presence of this symptom can be checked with the thumb test: in patients with arachnodactyly, part of the thumb (distal phalanx) protrudes beyond the edge of the clenched fist (Fig. 4).
The face of people with Marfan syndrome is usually elongated and thin. This is due to the high position of the upper palate, elongated skull and pathological slenderness.
They are also characterized by chest deformities, which can come in two varieties: inward (funnel-shaped chest) or outward (keeled chest, Figure 5).
Why should a doctor be consulted when detecting symptoms of Marfan syndrome?
The genetic abnormality itself is compatible with life. However, the consequences of the disease caused by the FBN1 mutation are dangerous:
- rupture of the large blood vessels, most commonly the aorta;
- Chronic heart failure - the inability of the heart to supply all organs with the necessary blood;
- Decreased visual acuity or complete loss of visual function.
Rupture of an aneurysm of the aorta or other large vessel often results in immediate death. Chronic heart failure can develop into an acute form and, without urgent medical treatment, can also have fatal consequences – sudden coronary death. It is these complications that most often lead to death in children with Marfan syndrome. A woman with FBN1 gene mutation syndrome is particularly at risk during pregnancy: the increased load on the aorta multiplies the risk of aortic rupture.
In order to prevent dangerous complications and compensate for the resulting disorders, parents should seek medical help as soon as possible at the first suspicion that their child has Marfan syndrome. It is important not only to examine the child once, but also to register with the doctors involved in correcting the symptoms:
- a specialist in genetics;
- a cardiologist
- a specialist in spinal orthopedics;
- a dermatologist
- ophthalmologist;
- gastroenterologist.
The list of specialists depends on the severity of the disease, and regular comprehensive preventive examinations allow early detection of new abnormalities.
Marfan Syndrome – a disease of geniuses?
Marfan syndrome is not only associated with many reasons for doctor visits. People with a mutation in the FBN1 gene often compensate for the physical symptoms of the disease with intellectual abilities, which is why this genetic disorder is also known as 'genius syndrome'. It is assumed that the increased adrenaline release as a result of pathological changes in the adrenal glands is responsible for the high mental and psychological activity of these patients. Therefore, there are also famous personalities among the people with Marfan syndrome. Julius Caesar, Abraham Lincoln and Charles de Gaulle, for example, were not prevented by pathology from becoming famous politicians; Hans Christian Andersen and Korney Chukovsky created exceptional literary works and Niccolò Paganini became famous as a brilliant musician.
Treatment of Marfan syndrome
Unfortunately, treatments for Marfan syndrome have not yet been developed. The therapeutic measures depend on the individual characteristics of the disease.
If the SCS disorders are not severe, conservative treatment is recommended. With significant expansion of the ascending aorta and aortic dissection, mitral valve prolapse, heart defects, surgical methods are used.
Vision is corrected with glasses and contact lenses. If necessary, surgical treatment of glaucoma, cataracts and replacement of the displaced lens with an artificial lens is performed.
To support the musculoskeletal system, collagen normalization therapy and a vitamin and mineral complex are prescribed. Severe skeletal abnormalities are surgically corrected.
Clinical guidelines exclude exercise, injury-prone games, and high activity levels for patients with the mutation. A corset can be used to support the spine, and physiotherapy and massage are recommended to strengthen the muscles.
Life is not easy for people with this gene mutation. However, by managing their health responsibly, patients can reduce the symptoms of the disease and prevent serious complications.
What Should Doctors Know About Marfan Syndrome?
Because it is a complex genetic condition, it is important for physicians to know and understand the main causes, symptoms, diagnostic principles and dangerous complications. These can be briefly summarized as follows:
- The cause is an autosomal dominant gene mutation with disturbed synthesis of fibrillin-1 in the body;
- the symptoms are different, the clinic is very different, but attention should always be paid to the patient's limbs, the condition of the lens and aorta;
- Complications also vary, but the most serious is aortic dissection;
- Marfan syndrome can only be accurately diagnosed by genetic testing.
Structural abnormalities of the skeleton, eyes and cardiovascular system are detected by diagnostic imaging, and drugs from the group of beta-blockers should be immediately prescribed to prevent aortic rupture.
Symptoms from different organs and systems
On the cardiovascular side, patients are most commonly diagnosed with valve prolapse and an aortic aneurysm. If the aortic root and the ascending part of the aorta are pathologically altered, this can lead to serious complications. Damage to the aortic intima most often occurs where the stress on the aorta is particularly high. There is a gradual widening of the vessel or a sudden rupture of the coronary sinus.
Aortic root dilatation is diagnosed in half of pediatric patients and approximately 60-80 % adult patients. Aortic regurgitation is diagnosed by auscultation, where a diastolic murmur can be heard over the valve. When the valve leaflets and cords are stretched, mitral valve prolapse can occur, accompanied by systolic clicks and murmurs of varying intensity. Bacterial endocarditis can occur in both the aortic and mitral valves.
On the musculoskeletal side, an important symptom is that the patient is tall and his arms are spread far above the height. On closer inspection, arachnodactyly of the thumb can be seen, with the distal phalanx protruding beyond the edge of the clenched fist. The thorax is either keeled and flared outwards or funnel-shaped and flared inwards. The joints are often hypermobile, with slight contractures in the elbows. Knees may be bent backwards, flat feet, and spinal curvatures such as kyphosis and scoliosis are common. The subcutaneous fatty tissue is also poorly developed.
Lens ectopia (subluxation or upward displacement) is the main visual sign of Marfan syndrome. Iridodenescence with fluttering of the iris is also common. When the lens is affected, its edge is displaced, which can be seen when the patient's pupils are not dilated. Myopia with spontaneous retinal detachment is also not uncommon.
In the area of the airways, pathological manifestations in the form of cystic lung disease and spontaneous pneumothorax, which usually recur frequently, often occur. As a result, patients often experience shortness of breath and chest pain.
Main signs of pathology

The external signs of Marfan syndrome often appear in the first few days after birth and only worsen later. Among the external signs suggesting pathology, the following should be mentioned first of all
- Increased length of the limbs and fingers (dolichostenomelia and arachnodactyly);
- underweight with increased physical development of the child;
- Elongated skull and face;
- Weak, underdeveloped muscle tissue, lack of adipose tissue;
- Unusually high joint mobility;
- Clumsiness and clumsiness in movement.
Marfan syndrome causes changes in the shape of the chest, curvature of the spine, and the development of flat feet in children over the age of four.
The most common ocular manifestations include myopia, ectopic lens, corneal changes, strabismus, iris and retinal hypoplasia. These changes often occur in the first few years of life, are bilateral, and progress systematically over time.
The most serious are the pathological changes in the cardiovascular system, which, if left untreated, can lead to death at a young age. These include changes in the vascular wall and various malformations of the heart and coronary arteries. In the worst form of the disease, a child will develop progressive heart failure within the first year of life.
In addition, the symptoms of Marfan syndrome can also show up in other systems and organs. The disease can affect the nervous tissue, bronchi and lungs, skin, urinary tract and genital tract.
Only a doctor can accurately diagnose the disease. Don't delay your consultation - call +7 (495) 775-73-60
How to accurately diagnose a disease in a child?
Currently, the diagnosis of Marfan syndrome is based on the clinical picture according to the Ghent criteria developed in 1995 and refined in 2010. These describe a number of signs of pathology of the skeletal system, organs of vision, heart and blood vessels, as well as other systems and organs. To determine the degree of compliance, the doctor collects the medical history (including family history), conducts a thorough examination of the patient using phenotypic tests, and orders laboratory and instrumental tests, which include:

- Urine analysis to determine glycosaminoglycans;
- DNA genotype detection;
- ECG and EchoCG to detect cardiac and vascular abnormalities;
- ultrasound examination of the heart;
- X-rays of the chest to detect malformations of the skeletal system, heart and lungs;
- Computed tomography and magnetic resonance imaging (MRI).
Additional tests and investigations may be arranged as needed.
Diagnosis of Marfan Syndrome
Diagnosing Marfan syndrome can be difficult because many patients present only certain characteristic signs and symptoms and no specific histological or biochemical changes. Because of this variability, diagnostic criteria are based on a combination of clinical findings, family history, and genetic history. (See the revised Ghent Nosology for more information on the diagnosis). However, the diagnosis in many individual cases of Marfan syndrome is unclear.
Homocystinuria Classic homocystinuria A series of defects in methionine metabolism leads to accumulation of homocysteine (and homocysteine dimers) with adverse effects such as a tendency to thrombosis, lens displacement and. Read more It can partially mimic Marfan syndrome but can be distinguished by detecting homocysteine in the urine. Genetic testing for mutations FBN1 can help diagnose in people who do not meet all clinical criteria, but there are also cases where the mutation in the FBN1 gene is negative FBN1 MUTATION. Prenatal diagnosis by mutation analysis FBN1 Gene is difficult to diagnose prenatally due to poor genotype/phenotype correlation (>1,700 different mutations have been described).
Standard skeletal, cardiovascular, and visual imaging studies are performed to detect clinically relevant structural abnormalities of critical diagnostic value (eg, echocardiography to detect aortic root dilatation).
In addition to the organ system criteria, a family history (relatives in the first knee with Marfan syndrome) and a genetic history (presence of FBN1 mutation causing Marfan syndrome) as the main criteria.
Prognosis in Marfan syndrome
Advances in therapy and regular monitoring have improved quality of life and reduced mortality. Life expectancy has increased from an average of 48 years in 1972 to near normal life expectancy for those receiving adequate medical care. However, the life expectancy of the average patient is still lower, mainly due to cardiac and vascular complications. This shortened life expectancy can be an emotional burden for young people and families.
Treatment of Marfan syndrome aims to prevent and treat complications.
In very tall girls, inducing precocious puberty at age 10 with estrogens and progesterone may limit growth in adulthood.
All patients should receive regular beta-blockers (eg, atenolol, propranolol) to prevent cardiovascular complications. These drugs decrease myocardial contractility and pulse pressure, reducing the progression of the aortic root aneurysm and the risk of aortic dissection. Angiotensin II receptor blockers can also be used.
If the aortic diameter is > 5 cm (smaller in children), prophylactic surgery is recommended. Pregnant women are at particular risk for aortic complications; recommended aortic reconstruction should be discussed before conception. Severe valve insufficiencies are also reconstructed surgically. Prophylaxis of bacterial endocarditis Infective endocarditis (IE) is an infection of the endocardium, usually caused by bacteria (more commonly streptococci or staphylococci) or fungi. It may present with fever, heart murmurs, and petechiae. Read more Not indicated before invasive procedures, except in patients who have had prosthetic heart valves implanted or who have infective endocarditis (Procedures requiring prophylactic antibiotic therapy in patients at high risk of endocarditis, used in the United States Procedures requiring prophylactic antibiotic therapy in patients at high risk of endocarditis, used in the United States. i Recommended prophylaxis against endocarditis during dental procedures and upper airway manipulation* Recommended prophylaxis against endocarditis during dental procedures and upper airway manipulation ).
What are the symptoms of Marfan Syndrome?

This disease is characterized by the fact that several systems are affected at the same time. It can be associated with a variety of clinical symptoms. The time of onset and the severity of symptoms can vary greatly from patient to patient.
- Above all, these people have certain external characteristics. They are characterized by their tall stature. Their torso is relatively short while their limbs are disproportionately long. The subcutaneous fat tissue is underdeveloped and the muscle tone is low. The facial skeleton is elongated and narrowed.
- It can also be associated with various skeletal deformities such as: B. a keel-shaped protrusion of the chest, a curvature of the spine and so on.
- The most striking feature of Marfan syndrome is damage to the cardiovascular system. Abnormal aortic lesions such as aortic aneurysms or heart valves are commonly found in these patients. Various congenital heart defects are possible. A common symptom is an abnormal heart rhythm.
- In severe cases, the heart malfunction can progress rapidly and lead to circulatory failure in a short time.
- In addition, eye disorders are often observed, which can manifest themselves in the form of myopia, lens ectopia, strabismus and others.
Other internal organs such as B. the respiratory system, can be affected. A common sign of airway involvement is spontaneous pneumothorax, a feature of Marfan syndrome, researchers at St. Petersburg State Pediatric Medical University demonstrated in a paper published in 2022.
How is Marfan syndrome treated?
No specific therapy has been developed for the treatment of Marfan syndrome as a whole. All treatments are symptomatic in nature.
First and foremost, measures are taken to restore and maintain normal cardiovascular function. In such patients, beta blockers, angiotensin converting enzyme inhibitors, etc. may be administered. If necessary, surgical procedures such as replacing the mitral valve with a prosthesis are performed.
Vision can be corrected by fitting glasses or by surgical procedures such as laser cataract removal.
In addition, preparations to increase collagen production, metabolic processes and vitamin complexes are used to increase collagen production.
How does Marfan syndrome manifest itself?
First of all, the special appearance of the patient is striking. People are tall, with long arms and legs out of proportion to other parts of the body, and thin, elongated fingers. The subcutaneous fat tissue is underdeveloped and muscle tone is reduced. The facial skeleton is long and narrow. A high arched palate and various occlusal anomalies are also characteristic.
Often patients with Marfan syndrome show excessive mobility of the joints and various structural changes in the spine and thorax.
There are always abnormalities in the structure of the heart and blood vessels. Aortic dilation, aortic aneurysms and various congenital heart defects such as B. a ventricular septal defect, can occur with this disease. The disease is often accompanied by cardiac arrhythmia.
Typically, Marfan syndrome is manifested by various eye abnormalities. In 2012, researchers at the Tashkent Pediatric Medical Institute conducted a study in which they found that eye damage occurs in 50-80 % patients, often as one of the first signs of the disease.
Clinical and morphological ocular abnormalities in Marfan syndrome include myopia, ectopic lens, underdevelopment of the iris, strabismus, etc.
In addition, other internal organs such as the central nervous system, bronchopulmonary system, skin, etc. can also be affected.
Treatment principles for Marfan syndrome

Due to the congenital character of Marfan syndrome, a complete cure is not possible. All therapy is symptomatic and is aimed more at stopping the progression of the disease.
To improve the cardiovascular system, beta blockers, angiotensin converting enzyme inhibitors, etc. may be prescribed. In the case of structural changes in the heart or blood vessels, surgical treatment, e.g. B. a reconstructive aortic surgery or a valve replacement can be performed.
Eye diseases can be corrected with glasses or contact lenses, surgery to remove glaucoma or cataracts, etc.
Major musculoskeletal changes may also require surgical procedures such as chest wall reconstruction, spinal stabilization surgery, etc.
Read more:- Marfan syndrome in newborns.
- Marfan syndrome.
- Marfan Syndrome - Summary.
- Ehlers-Danlo Syndrome.
- Treatment of short leg syndrome.
- How to correct a clubfoot in a child.
- Anatomy of the pelvic bones.
- ectrodactyly.