Abnormal changes in the body's main artery, the aorta, pose the greatest risk. 65-100 % of people with Marfan syndrome are at high risk of lesions in the bulb (the part of the aorta closest to the heart) and in the ascending arch of this artery, the parts that come directly from the heart. Because the inner layer of the vessel wall also contains connective tissue fibers, they are susceptible to wear and tear, and blood pressure in the aorta is higher than in other parts of the vascular bed. This causes the vessel to gradually expand and may cause abnormal blood pooling between the vessel walls, causing a sac-like bulge (aneurysm) or spontaneous rupture of the artery.

- Marfan syndrome
- First-class care: what modern diapers can do
- symptoms
- Causes of Marfan Syndrome
- Classification of Marfan syndrome
- How does Marfan syndrome manifest itself?
- Signs of Marfan syndrome
- Diagnostic methods for Marfan syndrome
- What is the prognosis for people with this disease?
- frequently asked Questions
- Causes of Marfan Syndrome
- How is it diagnosed?
- Phenotype and development of Marfan syndrome
- Treatment of Marfan syndrome
- How does Marfan syndrome manifest itself?
- Treatment options for Marfan syndrome
- Prognosis of Marfan syndrome
- Guidelines
Marfan syndrome
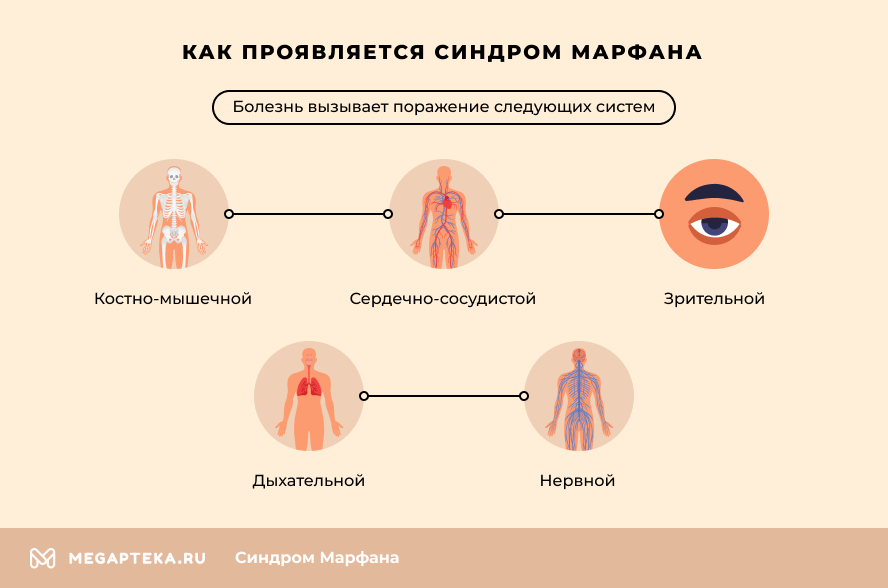
Marfan syndrome is an autosomal dominant systemic disease characterized by underdevelopment of connective tissue fibers due to structural defects in collagen. The most commonly affected organs are the visual organs, the musculoskeletal system, the heart and the blood vessels. The prognosis of the disease depends on the severity of the accompanying pathological changes. Most patients do not live past 50 years.
The characteristic symptoms of Marfan syndrome were first described by American doctors in 1875. The disease is named after the French pediatrician A. Marfan, who researched the disease for several years.
Today Marfan syndrome is not very common. The average incidence is between 1 in 20,000 and 1 in 5,000 people. This syndrome is neither racially determined nor does it have a racial pattern. This problem is neither racially determined nor is there a racial pattern.

First-class care: what modern diapers can do
symptoms
The symptoms of Marfan syndrome are varied.
Patients suffering from this disease tend to have a strange appearance. They are large, have a relatively short torso and excessively elongated limbs and upper arms. The patients' arms and legs are not only disproportionately long, but also very thin. The fingers are stretched and thin, the subcutaneous fatty tissue is underdeveloped and muscle tone is reduced.
The patient's head is long and narrow. There is often an anomaly of the maxilla with an upward curvature of the hard palate, resulting in a high arch. The jaw deformity results in an abnormal bite.
The patient's joints are very mobile (hypermobile) and the chest is deformed, with the sternum protruding forward (keel-shaped) or inward (funnel-shaped). In addition, there are various spinal deformities such as curvature in the anteroposterior plane.
Various pathological changes in the cardiovascular system are typical for Marfan syndrome. These can be represented by vascular wall defects such as aneurysms, valve and septal defects such as mitral valve prolapse, aortic root dilatation, etc. In 2013, researchers at the National Medical Research Center for Cardiovascular Surgery in Moscow, Russia announced the launch of a cardiac surgery program in 2013. AN Bakoulev Medical Center published the results of a study showing that aortic root dilation occurs in 60 % of patients with Marfan syndrome and mitral valve prolapse occurs in 91 % of patients.
In addition, the disease often affects the organs of vision. Clinically, it can manifest itself in the form of more or less severe myopia, lens ectopia, strabismus, corneal anomalies, etc.
Causes of Marfan Syndrome
The genetic disorder is caused by a defect in the FBN1 gene on the long arm of chromosome 15. This gene encodes the glycoprotein fibrillin-1, which is responsible for the strength and elasticity of connective tissue. Therefore, all manifestations of pathology are due to the fact that the connective tissue structures in the human body lose their normal properties.
The mutation is inherited in an autosomal dominant manner, meaning that children receive the pathological gene from parents who suffer from the pathology. There is a 50% chance that a child will receive the mutation from a parent (Figure 1). The syndrome is not inherited across generations: healthy children of affected parents cannot pass the gene on to their offspring.
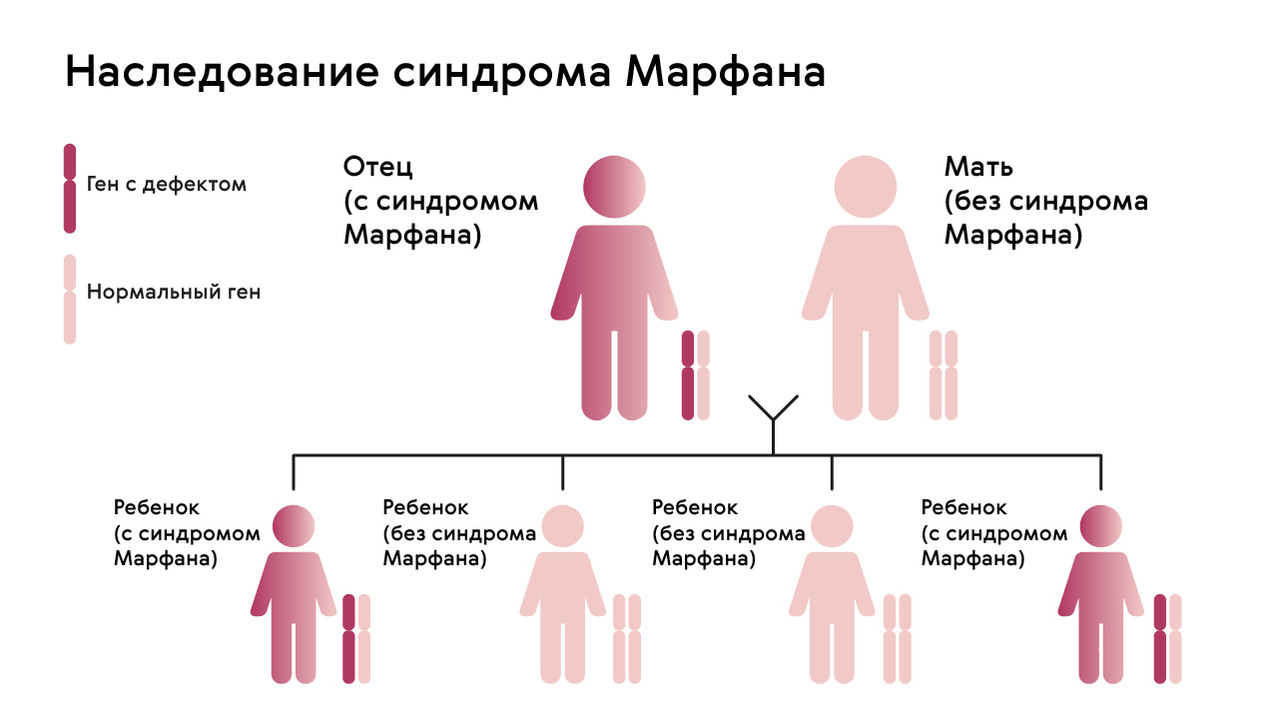
However, in about 25 % of people with Marfan syndrome, neither parent is a carrier of the FBN1 gene anomaly: in this case, the mutation develops spontaneously.
To date, no clear risk factors have been identified for this genetic disorder: the disease occurs equally in men and women, and its prevalence is independent of racial or ethnic group. The prevalence of this pathology is approximately 1 in 5-10,000.
If the clinical manifestations of the mutation are clear, the disease can be suspected already in the first months of life. However, mild forms of the disease often do not appear until adulthood, when the patient consults doctors about the various manifestations of the syndrome.
Important!!! Do not book an appointment for a genetic test as a medical examination. The search for a 'defect' in the FBN1 gene is only justified if the disease causes characteristic symptoms: asymptomatic inheritance of this mutation is not possible. If a parent has this diagnosis, the expectant mother should have a genetic test before birth. This makes it possible to determine in advance whether the anomaly has been transmitted to the child.
Classification of Marfan syndrome
Depending on the clinical manifestations of the genetic mutation, there are different forms of the disease.
There are two main clinical forms of pathology:
- Mild. These patients are 'lucky' because the abnormality affects only one or two body systems and their symptoms are mild. Despite the disease, those affected can lead an almost normal life.
- Severe. In these cases, three or more body systems are affected or one system is significantly impaired.
Depending on the degree of severity, there are mild, moderate and severe forms of Marfan syndrome. Severe pathologies are significantly rarer, with an incidence of around 1 in 25-50,000 people.
The nature of the course of the disease plays a decisive role in determining the prognosis of the disease:
- Progressive. In this case, new symptoms of the disease constantly appear, the severity of the disease increases, and the risk of fatal complications increases with each year of the patient's life.
- Stable. This clinical picture is considered the most favorable: in patients with stable symptoms of Marfan syndrome, the clinical picture remains largely unchanged throughout life.
Three different but similar diseases are distinguished:
- Marfan syndrome – a mild form of the disease with a positive genetic test.
- Marfan syndrome – the classic clinical picture with proven familial inheritance.
- Marfan syndrome – a manifestation of connective tissue pathology without a genetic mutation.
The first symptoms of the disease usually appear in childhood. When the patient reaches adolescence, disease progression becomes evident due to the mutation of the FBN1 gene.
How does Marfan syndrome manifest itself?
In the milder form, 1-2 body systems are affected (e.g. musculoskeletal and/or visual organs) and symptoms are mild. Despite the abnormalities, these patients lead almost normal lives. In the severe form, 3 or more body systems are affected, or there are significant changes in one of the systems.
The same genetic defect manifests itself in different ways, from mild changes to severe organ disorders.
- Musculoskeletal system – patients are characterized by tall stature, abnormal thinness and an elongated face. Other noticeable symptoms include elongated and thin fingers and irregular arm length. Posture is abnormal in most cases, with scoliosis or kyphosis. Patients often have flat feet and increased joint mobility.
- Cardiovascular system (CVS) – the syndrome causes cardiac arrhythmias, mitral valve insufficiency and various heart defects. Abnormal changes in the aorta are particularly dangerous. Most patients are at increased risk of dilation of the ascending limb and annulus and aneurysm formation.
- Vision – severe myopia, lens subluxation, or changes in lens position are most common. There is also an increased risk of retinal detachment. Glaucoma and cataracts often develop at a young age.
- Respiratory system – abnormal hypertrophy of connective tissue in the lungs leads to bronchoconstriction and pulmonary fibrosis. A genetic mutation often leads to the development of bronchial asthma.
- Nervous system – in most cases there are no abnormalities in the brain structures. However, an enlargement of the connective tissue capsule around the spinal cord leads to disturbances in leg movements, bladder and bowel function. A high release of adrenaline often leads to increased nervous excitability and hyperactivity.
Renal prolapse, uterine prolapse, varicose veins, and gastrointestinal (GI) abnormalities can also occur in Marfan syndrome.
Signs of Marfan syndrome
- Family history – an explanation for the parents' illness;
- Physical diagnosis – presence of typical symptoms, including the characteristics of anthropometric indices;
- Examination data – ECG (electrocardiography), EchoCG (echocardiography) of the heart, MRI (magnetic resonance imaging) of the brain and spine, X-rays, ophthalmological examination;
- Genetic testing.
Diagnostic methods for Marfan syndrome
Diagnosis of Marfan syndrome relies on clinical findings, genetic testing, and echocardiography, or MRI. Echocardiography and MRI detect abnormal changes in the aortic root and valves. Abnormal changes in the lens can be detected with a slit lamp, and enlargement of the dural sac in the spinal cord can be detected with MRI. When examining the eye, the ophthalmologist sees the typical sign of a genetic abnormality in the form of a lens subluxation.
However, in some cases it is difficult to make a correct diagnosis because many patients do not have the prominent symptoms of Marfan syndrome. They may also have no changes in biochemistry or histology. In such cases, doctors should focus on history taking, objective examination and genetic testing.
What is the prognosis for people with this disease?
Thanks to advances in modern medicine, the mortality rate for these patients is now significantly lower. While they used to live an average of no longer than 48 years, their life expectancy is now almost as long as that of 'normal' people. Of course, cardiac and vascular complications are still a problem.
- When prescribing beta blockers;
- the surgical correction of valvular and aortic anomalies;
- surgical correction of spinal anomalies.
Of the beta-blockers, it is advisable to prescribe propranolol or atenolol to patients. This prevents serious vascular and cardiac complications. Beta-blockers reduce the intensity of myocardial contraction, thereby reducing the load on the heart muscle, stopping the process of aortic dissection and reducing the risk of developing an aneurysm. If the aneurysm reaches a critical size, surgery is indicated.
Scoliosis is usually treated conservatively with spinal fixation, but if the spine is curved 40 degrees or more, surgery is preferred.
All patients with Marfan syndrome should be examined annually by a neurologist, a cardiologist and an ophthalmologist, with genetic counseling if necessary.
frequently asked Questions
Can I prevent my child from getting Marfan syndrome?
If Marfan syndrome runs in the family of at least one spouse, the couple should undergo genetic testing before conception. After pregnancy, prenatal screening is carried out: ultrasound of the fetus and a series of biochemical tests: maternal serum, amniotic fluid, chorionic biopsy, number of cells in the placenta and umbilical cord blood.
No. The diagnosis of Marfan syndrome in a conscript is a reason for dismissal from military service.
What preventative measures are there for Marfan syndrome?
Patients are monitored by a doctor throughout their lives. Preventive measures are aimed at preventing complications. For this purpose, cardiac surgical interventions are carried out, pharmacotherapy is carried out to eliminate the risk of vascular thrombosis, antibiotic therapy is carried out, etc.
Causes of Marfan Syndrome
The disease occurs in 1 in 10,000 people. Marfan syndrome is caused by a defect in a gene on chromosome 15 that determines the structure of fibrillin. This protein is the main component of elastin-associated microfibrils. The latter are found in most connective tissues, large blood vessels and ligaments that support the lens. The disease is inherited in an autosomal dominant manner: if one parent has the disease, there is a 50 % chance that the child will develop the pathology. There are cases when a healthy couple has a child with this genetic anomaly: a spontaneous mutation occurs in 1 out of 4 cases.
- keel or funnel curvature of the chest, spinal deformity
- disproportionate torso, tall stature
- long (curved) fingers (when the wrist is positive, the thumb and forefinger overlap when the other hand is tied)
- Increasing the range of motion of the limbs
- abnormal joint mobility
- Flatfoot Deformity
- weight loss
- bulging palate, tooth defects
- abnormal face (dolichocephaly, underdevelopment of the cheekbone, sunken eyes)
- Vision problems due to ectopia (displacement) of the lens, abnormal flattening of the cornea, nearsightedness, etc.
- Shortness of breath, weakness
- skin striae
- Recurrent hernias, etc.
How is it diagnosed?
The diagnostic algorithm depends on the systems involved. Consultations with the following specialists are required: geneticist, ophthalmologist, cardiologist, pulmonologist, gastroenterologist.
The instrumental study is based on:
- X-rays or computer scans of the hands, feet, chest, spine, hip bones, and skull.
- Magnetic Resonance Imaging (MRI) – when we want to diagnose changes in internal organs: anomalies of the viscera, heart defects, kidneys, etc. Magnetic resonance angiography of the chest allows a complete assessment of the arterial system.
- ECG, ultrasound, transthoracic echocardiography (EchoCG) of the heart and vessels;
Phenotype and development of Marfan syndrome
Marfan syndrome – is a multisystem disease with skeletal, ocular, cardiovascular, pulmonary, skin and other abnormalities. The skeletal anomalies include very high values (ratio of arm span to height >1.05; ratio of upper to lower segment
anomalies Eyes include lens subluxation, corneal flattening, eyeball elongation, and iris hypoplasia. Cardiovascular anomalies include mitral valve prolapse, aortic regurgitation, and ascending aortic aneurysm dilatation and dissection. Pulmonary abnormalities include spontaneous pneumothorax and alveolar enlargement. Cutaneous abnormalities include atrophic furrows and recurrent hernias. Dural anomalies include bulging of the dura in the sacral region.
Most Signs of Marfan syndrome occur with increasing age. Skeletal abnormalities, such as sternum anomalies and scoliosis, worsen as bones grow. Lens subluxation often occurs in early childhood but can also develop in adolescence.
With increased frequency Marfan syndrome Retinal detachment, glaucoma, and cataracts are common. Cardiovascular complications occur at any age and develop over the course of life.
The main causes of premature death are. Death of patients with Marfan syndrome are heart failure due to valvular insufficiency and aortic aneurysm and rupture. However, improvements in surgical and therapeutic care for aortic aneurysms have improved survival rates. From 1972 to 1993, the expected survival age for 50 % patients increased from 49 to 74 years in women and from 41 to 70 years in men.
abnormalities Phenotypic manifestations of Marfan syndrome:
– Age of onset: early childhood
– Disproportionately high
– Skeletal anomalies
– Ectopy (subluxation) of the lens
– Mitral valve prolapse
– Aortic aneurysm and rupture
– Spontaneous pneumothorax
– Hernia of the lumbosacral sheath
Treatment of Marfan syndrome
Marfan syndrome – is a clinical diagnosis made by the presence of specific symptoms. Confirmation of Marfan syndrome by detecting mutations in the FBN1 gene is currently not practical because the extreme heterogeneity of the alleles makes the identification of the causative mutation in each family extremely time-consuming and the correlation between genotype and phenotype is not reliable. Mutation analysis has low sensitivity and specificity for Marfan syndrome, limiting its clinical utility.
For Marfan syndrome there is no effective treatment; therefore, treatment focuses on prevention of complications and symptomatic treatment. Eye care includes regular checkups, correction of myopia, and often replacement of lenses. Orthopedic care consists of reinforcing treatment or surgical correction of scoliosis. Treatment of sternum abnormalities is primarily cosmetic.
physical therapy Can compensate for joint instability. Cardiovascular problems are treated through a combination of therapeutic and surgical measures. Therapeutic measures aim to prevent or delay aortic root dilatation by reducing cardiac parameters, reducing blood pressure and ventricular ejection force with beta-blockers, and limiting participation in contact sports, competitive sports, and isometric exercises.
Prophylactic replacement of the root Treatment for aortic regurgitation is indicated if the aortic dilatation or aortic regurgitation is severe enough. Most patients today are treated with supraclavicular aortic root replacement, which does not require continuous administration of anticoagulants.
Hemodynamics Pregnancy-related changes can lead to progressive aortic dilatation or dissection. Aortic dissection is thought to result from hormonal changes, increased blood volume, and increased cardiac output associated with pregnancy and childbirth. Current research suggests that the risk of pregnancy is too high if the width of the aortic root is more than 4 cm. Women may choose to have a supraclavicular aortic replacement before becoming pregnant.
How does Marfan syndrome manifest itself?
The first thing that stands out is the patient's special appearance. They are large, have long arms and legs disproportionate to other parts of the body, and thin and elongated fingers. The subcutaneous fatty tissue is underdeveloped and muscle tone is reduced. The facial skeleton is long and narrow. A high arched palate and various occlusion anomalies are also characteristic.
Often patients with Marfan syndrome show excessive mobility of the joints and various structural changes in the spine and thorax.
Abnormalities of the heart and blood vessels may also occur. The disease can present with aortic dilatation, aneurysms, and various congenital heart defects such as ventricular septal defect. The disease is often accompanied by cardiac arrhythmias.
Marfan syndrome typically presents with various eye abnormalities. In 2012, researchers at the Tashkent Pediatric Medical Institute conducted a study that showed that 50-80 % of patients experience visual impairment, often as one of the first symptoms of the disease.
The clinical and morphological eye abnormalities in Marfan syndrome may include myopia, ectopic lens, iris underdevelopment, strabismus, etc.
In addition, other internal organs such as the central nervous system, bronchopulmonary system, skin, etc. can also be affected.
Treatment options for Marfan syndrome

Due to the congenital nature of Marfan syndrome, there is no possibility of a complete cure. All treatment is symptomatic and largely focused on stopping disease progression.
To improve cardiovascular function, beta-blockers, angiotensin-converting enzyme inhibitors, etc. may be prescribed. Surgical treatment, e.g. B. Reconstructive aortic surgery or valve replacement may be performed if structural changes to the heart or vessels are evident.
The resulting eye diseases can be corrected with glasses or contact lenses and surgery to eliminate glaucoma, cataracts, etc.
Major musculoskeletal changes may also require surgical procedures such as chest wall reconstruction, spinal stabilization surgery, etc.
Prognosis of Marfan syndrome
Advances in therapy and regular monitoring have improved quality of life and reduced mortality. Life expectancy has increased from 48 years in 1972 to near-normal life expectancy for those who receive adequate medical care. However, the average life expectancy of patients is still lower, mainly due to cardiac and vascular complications. This reduced life expectancy can place emotional strain on youth and families.
Treatment for Marfan syndrome focuses on prevention and management of complications.
In very tall girls, induction of precocious puberty at age 10 with estrogens and progesterone may limit growth in adulthood.
All patients should receive regular beta-blockers (e.g. atenolol, propranolol) to prevent cardiovascular complications. These drugs reduce myocardial contractility and pulse pressure and reduce the progression of the aortic root aneurysm and the risk of aortic dissection. Angiotensin II receptor blockers can also be used.
If the aorta diameter is > 5 cm (smaller in children), prophylactic surgery is recommended. Pregnant women are at particular risk for aortic complications; recommended aortic reconstruction should be discussed before conception. Severe valve insufficiencies are also reconstructed surgically. Prophylaxis of bacterial endocarditis Infective endocarditis (IE) is an infection of the endocardium, usually caused by bacteria (more commonly by streptococci or staphylococci) or fungi. It can manifest itself with fever, heart murmurs and petechiae. Read more Not indicated before invasive procedures, except in patients with prosthetic heart valves or infective endocarditis (procedures requiring prophylactic antibiotic therapy in patients at high risk of endocarditis, US Recommended endocarditis prophylaxis for dental and upper airway procedures* Recommended endocarditis prophylaxis for dental and upper airway procedures ).
Guidelines
Marfan syndrome is caused by an autosomal dominant mutation of the gene encoding the glycoprotein fibrillin-1, which is a major component of microfibrils, so multiple deformities and defects are possible.
The manifestations are highly variable, but the main structural defects affect the cardiovascular and musculoskeletal systems as well as the visual system, resulting in the typical combination of limb lengthening, aortic root dilatation, and lens displacement.
To detect structural abnormalities, diagnostic imaging of the skeletal, cardiovascular, and ocular systems should be performed.
All patients should be treated with beta-blockers to prevent aortic complications; other complications should be treated as they arise.
Read more:- Marfan syndrome at a glance.
- Marfan syndrome in newborns.
- Marfan Syndrome - Summary.
- Ehlers-Danlo Syndrome.
- Treatment of short leg syndrome.
- Syndrome of the tibial nerve.
- tarsal and metatarsal joints.
- How to correct a clubfoot in a child.