The severity of the disease can vary within a family. [2]

- rubber man.
- History of the discovery of the syndrome
- introduction
- Causes of Ehlers-Danlos Syndrome
- Diagnosis of Ehlers-Danlos Syndrome
- Prognosis in Ehlers-Danlos Syndrome
- How Doctors Recognize Ehlers-Danlos Syndrome
- How Ehlers-Danlos Syndrome affects life
- Symptoms and Signs of Ehlers-Danlos Syndrome
- Complications of Ehlers-Danlos Syndrome
- Diagnosis of Ehlers-Danlos Syndrome
- Symptoms and Signs of Ehlers-Danlos Syndrome
- Complications of Ehlers-Danlos Syndrome
- Diagnosis of Ehlers-Danlos Syndrome
- The Ehlers-Danloh syndrome: a little history
- types of disease
- Connective tissue dysplasia: a current topic
- Connective tissue dysplasia: common clinical syndromes
- Valvular Syndrome
- to form
- Diagnosis of Ehlers-Danlos Syndrome
rubber man.

Ehlers-Danlos Syndrome (according to some sources Ehlers-Danlos Syndrome, EDS) is a rare inherited collagen pathology that affects the skin, musculoskeletal system and other organs. Other names for this syndrome are hyperelasticity of the skin, imperfect desmogenesis according to Rusakoff and Chernogubov-Ehlers-Danlos syndrome. People with this syndrome were seen in circuses in the past because they could surprise ordinary people with their looks and movements. Some famous people also had this syndrome, such as the violinist and virtuoso Niccolò Paganini.
History of the discovery of the syndrome
In the year 400 BC Hippocrates wrote about people with the disease in his work De aere, aquis, locis (On Air, Water and Places). He observed nomads and Scythians with weak joints and numerous scars all around them. Hippocrates believed the scars to be traces of chemical burns in an attempt to reduce hypermobility in the joints. Much later - in 1657 - the Dutch surgeon Jansun van Meekeren (1611-1666) described a Spanish boy with very elastic skin. The boy, named George Albes, could stretch the skin on his chin to his chest and the skin on his knees to mid-shin, but only on the right side of his body. The world-famous violinist and composer Niccolò Paganini (1782-1840) had hypermobile joints, a slim build and a deformed chest - all symptoms typical of a carrier of Ehlers-Danlos Syndrome. Towards the end of the 19th century, some SED patients appeared in traveling exhibitions as people with unusual abilities – such as the 'flexible lady' described by the American doctors George M. Gould and Walter L. Pyle. For a long time, the variety of clinical manifestations of the syndrome prevented a detailed description of all forms of the disease.
The first detailed description of SED was presented by Andrey Chernogubov, a Russian doctor at Moscow's Myasnitskaya Hospital, at a meeting of the Moscow Society of Venereology and Dermatology in 1892. He described two patients with increased mobility of the large joints. One was a 17-year-old boy with epilepsy whose skin was fragile and hyperelastic and unable to hold sutures.
Later, in 1901, the Danish dermatologist Edvard Lauritz Ehlers published the description of a patient with weak joints and hyperelastic skin and a tendency to bruise. At a meeting of the Danish Dermatological Society in 1899, he demonstrated this disease. Seven years later, the French doctor Henri-Alexandre Danlos examined a patient with vascular lesions on the elbows and knees. Individual descriptions of the syndrome have since appeared in both the United States and England, and by 1966 the total number of reports rose to 300. In 1936, the English dermatologist Frederick Parkes Weber associated all cases with hyperelasticity and fragility of the skin and hypermobility of the joints . He called the new disease Ehlers-Danlos syndrome.
introduction
Ehlers-Danlos syndrome (imperfect desmogenesis, hyperelastic skin) is one of the hereditary collagenopathies along with osteogenesis imperfecta, Marfan syndrome and others. Ehlers-Danlos syndrome is heterogeneous and includes a heterogeneous group of hereditary connective tissue lesions (connective tissue dysplasia) associated with impaired biosynthesis of collagen proteins. The symptoms of Ehlers-Danlos syndrome are systemic and affect the musculoskeletal system, the skin, the cardiovascular system, the ocular system and the dental system, among others. The Ehlers-Danlos syndrome is therefore of practical interest not only for genetics, but also for traumatology and orthopedics, dermatology, cardiology, ophthalmology and dentistry.
Difficult verification and the presence of light forms make it difficult to obtain accurate information about the real frequency of Ehlers-Danlos syndrome; the diagnosis rate of mild cases is 1 in 5 000 in newborns, while the frequency of severe cases is 1 in 100 000.
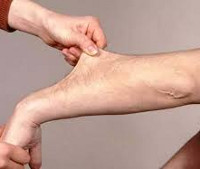
Causes of Ehlers-Danlos Syndrome
The different forms of Ehlers-Danlos syndrome differ in the mode of inheritance and the primary molecular and biochemical defects. However, all clinical forms are due to gene mutations that are responsible for quantitative or structural abnormalities in collagen. To date, the molecular mechanisms of the Ehlers-Danlos syndrome have not been clarified for all forms of the disease.
For example, it is known that type I syndrome is characterized by reduced fibroblast activity, increased synthesis of proteoglycans and a deficiency in enzymes responsible for normal collagen biosynthesis. Ehlers-Danlos syndrome type IV is associated with insufficient production of type III collagen, and type VI is characterized by a deficiency in the enzyme lysyl hydroxylase, which is involved in the hydroxylation of lysine in procollagen molecules. Type VII is caused by abnormalities in the conversion of Type I procollagen to collagen; Type X due to abnormalities in plasma fibronectin involved in the organization of the intercellular matrix, etc.
The pathomorphological picture in the different types of Ehlers-Danlos syndrome is characterized by thinning of the dermis, confusion and loss of compactness of collagen fibers, hypertrophy of elastic fibers, an increased number of vessels and their lumen.
Diagnosis of Ehlers-Danlos Syndrome
Doctors diagnose Ehlers-Danlos syndrome based on symptoms and physical examination. Genetic testing is usually done to confirm the diagnosis.
Doctors can try to identify certain types of Ehlers-Danlos syndrome by taking a sample of skin for examination under a microscope (biopsy).
Other tests are done to look for possible diseases that have associated complications. For example, echocardiography Echocardiography and other ultrasound procedures An ultrasound scan uses high-frequency waves (ultrasound). The waves reflected from the internal structures are recorded with a moving image. X-rays in. Read more information and other imaging tests to detect heart or blood vessel problems.
Prognosis in Ehlers-Danlos Syndrome
Despite the numerous and varied complications, the life expectancy of patients with Ehlers-Danlos syndrome is generally normal. However, in some people with certain forms of Ehlers-Danlos syndrome, the complications (usually bleeding) are fatal.
It is not possible to completely cure Ehlers-Danlos syndrome or to correct connective tissue abnormalities. Injuries can be treated, but this can be difficult for the doctor because suturing the incisions can tear delicate tissue. Incisions are usually easier to close with tape or medical grade skin glue and leave fewer scars.
Special precautions should be taken to avoid injury. For example, children with severe Ehlers-Danlos syndrome should wear protective clothing and shields.
Operations require special techniques to minimize trauma and ensure a large supply of blood for transfusion. The obstetrician (a doctor who specializes in childbirth and the care and treatment of pregnant women) should monitor the progress of the pregnancy and delivery. Genetic counseling for family members is recommended.
Copyright © 2023 Merck & Co, Inc, Rahway, NJ, USA and its subsidiaries. All rights reserved.
How Doctors Recognize Ehlers-Danlos Syndrome
The Cleveland Clinic, a large private medical center in the US, explains how doctors diagnose EDS in patients:
- Genetic testing is the most common way to identify a faulty gene.
- A biopsy is a test in which a doctor takes a sample of skin and examines it under a microscope to look for signs of disease, such as certain genes and gene mutations (abnormalities).
- A physical exam where the doctor determines how much the skin is stretched and how much movement the joints have.
- X-rays and CT scans provide images of internal parts of the body to detect abnormalities, heart problems, and abnormal bones.
In Russia, SED diagnosis is made on the basis of a visual examination and EMG electromyography, which assesses the condition of the skeletal muscles and nerves. To determine the type of SED syndrome, tests for types of collagen can be performed: many private clinics offer tests to detect this syndrome.
How Ehlers-Danlos Syndrome affects life
Ehlers-Danlos syndrome affects sufferers in different ways throughout life. Other diseases can be diagnosed: neurological, spinal or cardiovascular diseases. For example, in the vascular form of the syndrome, blood vessels can rupture, which in turn can cause internal bleeding or stroke. People with this type also have an increased risk of organ rupture.
- joint and muscle pain, which can sometimes be very severe;
- weakening of connective tissue, which can lead to hernias;
- debilitating chronic fatigue that negatively affects mental and physical abilities;
- cardiovascular disorders
- gastrointestinal disorders;
- gynecological problems.
In addition, a person with SED may experience dizziness, memory and attention problems, altered sweating, and mental health problems.
Symptoms and Signs of Ehlers-Danlos Syndrome
The symptoms and signs of Ehlers-Danlos syndrome vary widely.
The predominant symptoms are hypermobility of the joints, abnormal scarring and impaired wound healing, brittle blood vessels and velvety, over-extensible skin. The skin can be stretched several centimeters, but returns to its normal state after relief.
Broad, parchment-like scars often cover bony prominences, particularly on the elbows, knees, and lower legs; in the hypermobile type, the scars are less pronounced. Soft pseudotumors (fleshy growths) often form on the surface of scars or bruises.
The degree of joint hypermobility varies but can be significant in arthrochalasia, the classic and the hypermobile forms.
Bleeding tendencies are rare, although the vascular type is characterized by vessel rupture and bruising. Subcutaneous calcified globules may be palpable or visible on x-rays.
Complications of Ehlers-Danlos Syndrome
Minor trauma can result in a wide open wound that bleeds little; surgical closure of wounds can be difficult as sutures tend to tear on fragile tissue. Surgical complications arise due to the fragility of the deep tissue. The sclera can be very fragile, which can lead to kyphoscoliosis-like ocular perforations.
Moderate synovial effusions, ligament sprains, and dislocations are quite common. Vertebral kyphoscoliosis develops in 25 % of the patients (especially in patients with kyphoscoliosis type), thoracic deformities in 20 % and equinovarus clubfoot foot deformity (clubfoot) and other foot abnormalities of the foot) and forefoot adduction (medial deviation from the vertical axis of the foot). Read more information – under 5%. About 90 % of affected adults have flat feet. Equinovarus clubfoot is characterized by plantar flexion, heel inward tilt (from the midline of the foot), and forefoot adduction (medial deviation from the vertical axis of the foot). Read more . Progressive hip dysplasia Progressive hip dysplasia (PDTS) Progressive hip dysplasia (formerly congenital hip dislocation) is a developmental anomaly of the hip joint. (See also Introduction to Congenital Craniofacial and Musculoskeletal Dysplasia. Read more (previously Congenital Hip Dislocation) occurs in 1 % of cases (the arthrochalasic type is characterized by bilateral progressive hip dysplasia).
Diagnosis of Ehlers-Danlos Syndrome
Echocardiography and/or other vascular studies to detect cardiovascular complications
The initial diagnosis of Ellens-Danlos syndrome is based primarily on clinical findings but must be confirmed by genetic testing, which is now able to identify most subtypes. Ultrastructural evaluation of skin biopsy specimens can help diagnose the classic, hypermobile, and vascular types.
Echocardiography and other imaging tests of the vessels are done to detect heart disorders (eg, prolapsed valves, aneurysms), which can occur in some types.
Symptoms and Signs of Ehlers-Danlos Syndrome
The symptoms and signs of Ehlers-Danlos syndrome vary widely.
The predominant symptoms are hypermobility of the joints, abnormal scar formation and impaired wound healing, fragile blood vessels and velvety, overstretched skin. The skin can be stretched several centimeters, but returns to its normal state after relief.
Broad, parchment-like scars often cover bony prominences, particularly on the elbows, knees, and lower legs; in the hypermobile type, the scars are less pronounced. Soft pseudotumors (fleshy growths) often form on the surface of scars or bruises.
The degree of joint hypermobility varies but can be significant in arthrochalasia, the classic and the hypermobile forms.
Bleeding tendencies are rare, although the vascular type is characterized by vessel rupture and bruising. Subcutaneous calcified globules may be palpable or visible on x-rays.
Complications of Ehlers-Danlos Syndrome
A small trauma can result in a wide open wound that bleeds little; surgical wound closure can be difficult because sutures tend to tear on fragile tissue. Surgical complications arise due to the fragility of the deep tissue. The sclera can be very fragile, which can lead to kyphoscoliosis-like ocular perforations.
Moderate synovial effusions, ligament sprains, and dislocations are quite common. Vertebral kyphoscoliosis develops in 25 % of patients (particularly in patients with kyphoscoliosis type), thoracic deformities in 20 % and equinovarus clubfoot foot deformity (clubfoot) and other foot abnormalities. from the midline of the foot) and forefoot adduction (medial deviation from the vertical axis of the foot). Read more information – on page 5. Approximately 90 % of affected adults have flat feet. Equinovarus clubfoot is characterized by plantar flexion, inward heel tilt (from the midline of the foot), and forefoot adduction (medial deviation from the vertical axis of the foot). Read more . Progressive hip dysplasia Progressive hip dysplasia (PDTS) Progressive hip dysplasia (formerly congenital hip dislocation) is a developmental anomaly of the hip joint. (See also Introduction to Congenital Craniofacial and Musculoskeletal Dysplasia. Read more (previously Congenital Hip Dislocation) occurs in 1 % of cases (the arthrochalasic type is characterized by bilateral progressive hip dysplasia).
Diagnosis of Ehlers-Danlos Syndrome
Echocardiography and/or other vascular studies to detect cardiovascular complications
The initial diagnosis of Ellens-Danloh syndrome is based primarily on clinical findings, but must be confirmed by genetic testing, which can now identify most subtypes. Ultrastructural evaluation of skin biopsy specimens can help diagnose the classic, hypermobile, and vascular types.
Echocardiography and other imaging tests of the vessels are done to detect heart disorders (eg, prolapsed valves, aneurysms), which can occur in some types.
The Ehlers-Danloh syndrome: a little history
The disease was first mentioned in the writings of Hippocrates, who died in 400 BC. described nomads with weak joints. The arms were treated by cauterizing the joints to scar them with burns, thus minimizing joint hypermobility.
Another doctor was a Dutch surgeon, JJK Van Meekeren, who described a Spanish boy with incredibly elastic skin. The child could stretch his skin from his knees to halfway up his shins, and the skin from his chin easily reached halfway up his chest. It is true that only the skin on the right side of the boy's body showed such remarkable elasticity.
The first detailed description of the disease came from Andrei Chernogubov, a Russian dermatologist who worked at Moscow's Myasnitskaya Hospital in the late 19th century. The doctor presented descriptions of two patients at a meeting of the Moscow Society of Venereology and Dermatology in 1892. In 1901 the Dane Edward Ehlers published the description of a patient with hyperelastic skin, weak joints and a high tendency to bruise. Seven years later, another patient was described by French dermatologist Henri Danleau. By 1966, the number of cases described had risen to 300. But as early as 1936, the English dermatologist Frederick Weber had systematized the existing descriptions and combined cases of joint hypermobility, skin fragility and hyperelasticity into a single clinical picture, which he called Ehlers-Danlos syndrome. Apparently Weber was unaware of the Chernogubow syndrome or ignored its primacy. Today there are several variants of the name of the disease. For the sake of brevity, in the remainder of this article, we will refer to the most common abbreviation – SED. However, we should not forget that the first in the new history was still Chernogubov.
types of disease
SED is an inherited disease in which the molecular structure of collagen is disrupted. It affects more than just the properties of the skin. By the end of the 20th century, scientists had already identified six main types of the disease:
- classic;
- hypermobile
- vascular;
- kyphoscoliosis;
- achondroplasia;
- dermatosparaxis.
All are linked to a defect in genes that affect collagen synthesis, but they occur in different ways and with different frequencies. 'Faulty' genes cause the collagen fibers to lose their characteristic structure. Mutations can occur randomly, but there are also known cases of familial SED. In particular, in the Azerbaijani village of Gobu in Apsheron district, the birth rate of children with SED is much higher than in other areas, around 10 %.
- Classical SED is not the most common form of the disease - for every 20-40 thousand people there is one patient with hyperextracted skin, fragile blood vessels that bruise easily, and wounds that heal with the formation of atypical scars and scarring. Subcutaneous cysts and benign tumors are also common.
- Hypermobile EDM is more common: one case per 10,000 to 15,000 people. The main symptom is the weakness of the small and large joints, easy dislocation and the early development of osteoporosis. Gastrointestinal disorders such as intestinal discomfort, gastroesophageal reflux, and gastritis are also characteristic of this form.
- The vascular form of SED is less common: one case in 250,000 people. Due to the fragility of the blood vessels, severe bleeding, both external and internal, is common. For this reason, the life expectancy of these patients rarely exceeds 50 years. In this form there is no pronounced hyperelasticity of the skin.
- The achondroplasmic type is even rarer: only 30 cases have been described. It is characterized by a congenital dislocation of the hip, a tendency to bruise and scar, a predisposition to early arthritis, and very elastic skin.
Connective tissue dysplasia: a current topic
The term 'dysplasia' refers to a failure in the formation of a specific structure. In our case it is the connective tissue. It is a congenital condition in which there are mutations in the genes responsible for the development of connective tissue components.
This pathology is widespread. So, according to statistics, in Russia one in 10 residents is diagnosed with connective tissue dysplasia. According to various sources, 10-70 % of the world's population have this diagnosis.
This pathology is difficult to diagnose - for many years the patient can consult specialists of different specialties with different ailments, without getting an unequivocal diagnosis.
Connective tissue dysplasia: common clinical syndromes

Connective tissue dysplasia is a disease with a variety of clinical manifestations, the severity of which depends on several factors:
- the type and number of mutations
- the type of tissue affected (loose or dense connective tissue);
- the nature of the anomaly in the phase of connective tissue formation.
Because connective tissue is an integral part of many organs and systems, the disease is multisystemic. It can take a different course in each patient and affect more or less specific organs. This results in a diverse clinical picture.
In medicine, a distinction is usually made between the forms of connective tissue dysplasia:
The most severe symptoms, which occur in constant combination, are divided into different syndromes. There are different forms of dysplasia:
These syndromes are quite rare, the clinical picture of these diseases is striking and the diagnosis is usually unequivocal.
Undifferentiated dysplasia, which does not fit into the clinical picture of one of the differentiated syndromes, is problematic. The presence of these features, which do not give a clear clinical picture but cause concern for patients and affect their quality of life, is called undifferentiated connective tissue dysplasia.
The interesting thing is that every patient is unique. There are so to speak no 'standard' symptoms and signs for this disease. Therefore, as a rule, several syndromes are diagnosed, based on the dominance of one system or another.
Valvular Syndrome
Changes in the heart valve apparatus are diagnosed in 'dysplastic children' as early as childhood. The most common diagnosis is mitral valve prolapse. Lesions can also affect the aortic and tricuspid valves, but are less common.
to form
The Ehlers-Danlos syndromes comprise a heterogeneous group of disorders characterized by soft tissue fragility and widespread manifestations in the skin, ligaments and joints, blood vessels and internal organs. The clinical spectrum ranges from mild hyperelasticity of the skin and joints to severe physical disabilities and life-threatening vascular complications.
Originally, the 11 forms of Ehlers-Danlos syndrome were named with Roman numerals (Type I, Type II, etc.). In 1997, researchers proposed a simpler classification (Villefranche nomenclature) that reduced the number of types to six and gave them descriptive names based on their main characteristics. [7]
The current Villefranche classification recognizes six subtypes, most of which are associated with mutations in one of the genes encoding collagenous fibril proteins or enzymes involved in the post-translational modification of these proteins. [8th]
- Classic Type I (OMIM 606408)
- Type II Classic type, Ehlers-Danlos syndrome with tenascin X deficiency
- Type III hypermobility type
- Type VIA, Type VIB Vascular type (OMIM 225320)
- Type VIIA and VIIB Arthrochalasia type (OMIM 130060, 617821), Type VIIC Dermatosparaxis (OMIM 225410), Progeroid type
- Type VIII periodontitis type, Ehlers-Danlos variant with periacetabular heterotopia
Determining the correct EDS subtype has important implications for genetic counseling and treatment, and is favored by specific biochemical and molecular testing. [9]
Diagnosis of Ehlers-Danlos Syndrome
The scope of the examination depends on the presence of the main clinical features of the disease. Genealogical studies and molecular genetic methods are required.
The following prerequisites are necessary for the diagnosis of Ehlers-Danloh syndrome.
- The presence of at least one major criterion is required for clinical diagnosis. With sufficient capacity, the presence of one or more main criteria guarantees confirmation of Ehlers-Danloh syndrome at the laboratory level.
- A minor criterion is a feature that demonstrates a lower level of diagnostic specificity. The presence of one or more minor criteria contributes to the diagnosis of a particular type of Ehlers-Danlos syndrome.
- If there are no major criteria, minor criteria are not sufficient to make the diagnosis. The presence of minor criteria indicates the presence of a condition similar to Ehlers-Danlos syndrome, the nature of which will become clearer as its molecular basis becomes known. Since the prevalence of the minor criteria is much higher than that of the major criteria, the presence of only minor criteria forms the basis for the diagnosis of an Ehlers-like phenotype, in full agreement with the Wilfranche revision.
The diagnosis of the classic syndrome is made in a patient based on the minimal clinical diagnostic criteria (hyperelasticity of the skin and the presence of atrophic scars) and the identification of the pathogenic COL5A1, COL5A2 or COL1A1 gene by molecular genetic testing.
Hypermobility of the joints is also one of the diagnostic criteria for Morfan syndrome and Ehlers-Danloh syndrome. If the appropriate criteria are not met, hypermobility should be treated as a separate condition.
Read more:- tarsal and metatarsal joints.
- Marfan syndrome in newborns.
- Marfan syndrome at a glance.
- Marfan syndrome.
- Marfan Syndrome - Summary.
- Treatment of short leg syndrome.
- extension and flexion of the foot.
- tarsus.