Charcot-Marie-Tooth neuromyotrophy (CMT) refers to a group of progressive, chronic, inherited polyneuropathies that include Roussi-Levy syndrome, hypertrophic degenerative Sott neuropathy, Refsum disease and other, rarer diseases.

- Neural atheropathy: causes, symptoms, diagnosis
- International Classification of Neural Atheropathies
- Background information.
- Causes of Roussi-Levi Syndrome
- causes
- pathogenesis
- Symptoms of Refsum disease
- diagnosis
- Pathogenetic aspects of Charcot-Marie-Tooth disease and its symptoms
- Classification of Charcot-Marie-Tooth disease
- Effect
- frequently asked Questions
- classification
- diagnosis
- clinical picture
- diagnosis
- Treatment approach
- prevention
Neural atheropathy: causes, symptoms, diagnosis
Syndromes of demyelination of nerve fibers with autosomal dominant inheritance through the X chromosome occur in 2 people per hundred thousand inhabitants. The disease is not typical for the territory of the Russian Federation, since there is no reservoir for the existence of pathogens on the domestic territory.
The chronic course of the disease is accompanied by a gradual progression of muscle paralysis and paralysis. The pathology is accompanied by changes in the muscles, which gradually atrophy first in the legs and secondarily in the arms. Fasciculations are caused by disruption of cholinesterase transmission of nerve fibers as a result of an infectious lesion. Supportive treatment is aimed at restoring antioxidant function and metabolic dysfunction.
The reduced nutrient intake is characterized by atrophic changes in the muscles and loss of contractile function. The pathogenetic mechanisms are due to the destruction of the motor neurons of the spinal cord, trunk structures and the frontal horns of the midbrain.

Multifocal leukoencephalopathy on MRI
International Classification of Neural Atheropathies
A number of pathologies associated with motor and sensory impairments are classified as hereditary amyotrophies. The international classification ICD 10 codes this nosology as 'G12'. Heredity prevents etiological treatment. A syndrome of common disorders with deafness, retinitis pigmentosa, and ataxia is treated with supportive medications.
- Charcot-Marie-Tooth;
- Dejerin-Sotta – hypertrophic lesions of nerve fiber sheaths cause a gradual increase in clinical symptoms due to sensorimotor neuropathy;
- Degenerative changes in the trunk, horns of the spinal cord occur in three clinical forms - late, early and congenital. The nosology is referred to as 'Werdnig-Hoffmann spinal muscular atrophy';
- The destruction of motor neurons with subsequent spinal horn atrophy is characterized by muscle twitching. This pathology is called 'Kullberg-Wehlander atrophy';
- Other hereditary amyotrophies include neurogenic malignant, scapulohumeral and New England amyotrophy.
According to the International Classification of Diseases, symptoms with similar clinical features should be distinguished. Related manifestations are treated according to different patterns, so a differential diagnosis is necessary.
- The first type involves altered properties of nerve signaling amid destruction of myelin sheaths;
- In the second variant, impulsivity persists, but axonal damage to the Schwann cells is detected.
According to Wikipedia, early verification of the morphological forms of the disease is necessary before complications develop.
Background information.
Roussi-Levy syndrome is named after the French neurologist Levy and the pathologist Roussay, who described it in 1926. The authors described the syndrome as familial areflextic dysstasis with slow progression. Ataxia and tendon aflexia are the primary clinical manifestations of the syndrome, which gives rise to its other name: ataxia-areflexia syndrome. According to the 10th revision of the international classification, Russi-Levi syndrome is classified in the group of hereditary motor-sensory neuropathies (HMSN). However, there is no clear consensus among clinicians on how to classify this pathology. According to the symptoms, some neurologists classify Roussi-Levy syndrome as a clinical form that lies between Charcot-Marie-Tooth disease and Friedreich ataxia; others consider it a separate clinical variant of spinal amyotrophy. Despite the genetic evidence supporting the latter statement, many neurologists still treat Roussi-Levy syndrome as a separate nosology.
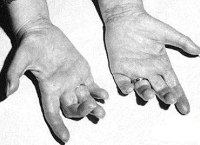
Causes of Roussi-Levi Syndrome
The familial occurrence of the disease suggests a hereditary origin. Roussi-Levy neuropathy has previously been found in families with recurrent cases of Charcot-Marie-Tooth disease. Molecular genetic studies carried out in recent years have revealed that the genetic basis of the syndrome is an anomaly on chromosome 17 (locus 17p11.2) in the form of duplications of individual genes and point mutations in the myelin protein null gene (MPZ). Based on this finding, Roussi-Levi syndrome can be viewed as a phenotypic form of Charcot-Marie-Tooth neuronal amyotrophy (NMSN variant IA).
This syndrome is inherited in an autosomal dominant manner. Morphologically, degenerative changes in the posterior spinal roots and peripheral nerve trunks are evident in the form of moderate demyelination and reduction of nerve fibers; dystrophic changes in the pyramidal and cerebellar tracts running in the lateral columns of the spine; Degeneration of the posterior columns and atrophy of the muscle fibers of the skeletal muscles.
causes
To date, practical neurology as a science does not have reliable data on the etiology and pathogenesis of nerve amyotrophy. Studies have shown that 70-80% of CMT patients who have undergone genetic testing have a duplication of a specific region of chromosome 17. Charcot-Marie-Tooth neuronal amyotrophy can take various forms, possibly due to mutations in different genes. Researchers found that the form of CMT caused by a mutation in the gene for the mitochondrial protein MFN2 leads to clumping of the mitochondria, which disrupts their movement along the axon.
Charcot-Marie-Tooth disease is characterized by an autosomal dominant inheritance pattern with a penetration rate of 83 %. There are also cases of autosomal recessive inheritance.
pathogenesis
In most forms of CMT, damage to the myelin sheath of peripheral nerve fibers has been noted; Less commonly, the axons, the axial cylinders that run through the center of the nerve fiber, are affected. Degenerative changes also affect the anterior and posterior spinal cord roots, the anterior horn neurons, the Gaul's tracts (spinal deep sensory pathways) and the Clark's columns, which are part of the posterior tracts of the spinal cord.
Secondarily, as a result of the dysfunction of the peripheral nerves, muscle atrophy develops, in which certain groups of myofibrils are involved. Further progression of the disease is characterized by displacement of the sarcolemmal nuclei, hyalinization of the altered myofibrils and hypertrophy of the interstitial connective tissue. As a result, increasing hyaline degeneration of the myofibrils leads to their collapse.
Symptoms of Refsum disease
Multiple lesions of the peripheral nerve trunks lead to the symptoms of polyneuropathy: numbness in the hands and feet, paresthesia, reduced pain perception and deep (proprioceptive, vibratory) sensitivity. Polyneuropathy is caused by sensorimotor factors. The motor disorders begin mainly with paresis of the extensor muscles of the feet, then the flaccid paresis spreads to the distal parts of the legs and arms, accompanied by hypotrophy of the muscles of the feet, lower legs, hands and forearms. Cerebellar ataxia with unsteady gait, hypermetria and altered handwriting are characteristic. In vision, reduced visual acuity, hemeralopia, narrowed visual fields and, in some cases, cataracts are observed. Sometimes photophobia is present.
Refsum disease is often accompanied by hearing loss, loss of smell (anosmia), drooping upper eyelids and oculomotor disorders. The skin is dry and flaky. The severity of ichthyosis varies from mild hyperkeratosis of the feet and hands with small white crusts to severe lamellar ichthyosis. Some patients have accompanying skeletal dysplasias: curvature of the spine (scoliosis), foot deformities (cavus foot, curvature and shortening of the toes).
diagnosis
The diagnosis can be made using a blood test and a urine test that determine the concentration of phytanic acid. This value increases to 800 µmol/l in Refsum disease, while the normal range is below 19 µmol/l. An ophthalmologist is consulted for all patients. Examination of visual acuity shows reduced visual acuity, perimetry shows concentric narrowing of the visual fields, and ophthalmoscopy shows signs of optic nerve atrophy and polymorphic degenerative changes in the retina.
Electroneuromyography can determine the polyneuropathic nature of the peripheral nervous system and reveal anomalies of the anterior horn motoneurons. Elevated albumin levels are diagnosed in the CSF. Disorders of the sense of smell are determined using olfactometry. Hearing disorders are diagnosed through consultation with an audiologist and through audiometry.
The presence of bone deformities is the basis for prescribing an X-ray of the spine, CT or X-ray of the feet, followed by a consultation with an orthopedist. To evaluate the heart, an EKG is performed and a cardiologist is consulted. A genealogical study is carried out to determine the type of inheritance of the pathology. The final diagnosis can be confirmed by DNA analysis.
When diagnosing Refsum disease, it is distinguished from Charcot-Marie-Tooth amyotrophy, polyneuropathies of other origins (Degenerina-Sotta syndrome, diabetic neuropathy, Russ-Levy syndrome, paraneoplastic neuropathy, etc.) and Friedreich ataxia.
Pathogenetic aspects of Charcot-Marie-Tooth disease and its symptoms
The pathogenesis of Charcot-Marie-Tooth disease is due to nerve damage leading to secondary muscle atrophy. The fast-moving motor nerves that supply the most heavily used muscles in the limbs - the foot and lower leg muscles - are most often damaged. The muscles of the hands and forearms are affected slightly less and later. The sensory nerves are also affected, causing a loss of sensitivity to touch, pain and temperature in the limbs.
Charcot-Marie-Tooth disease usually manifests itself between the 10th and 20th. Initially there is a symmetrical weakness of the lower limbs, which manifests itself in a characteristic step or a so-called 'rolling gait'. Then the feet begin to twist and deform, the arch of the foot widens and a hollow foot develops. As muscle loss progresses, the feet take on the appearance of upside-down bottles.
The hands are gradually also affected: the atrophy of the hand makes it look like a monkey's paw. In addition, sensitivity, especially superficial sensation, is impaired. Sometimes there is cyanosis of the affected limbs and swelling of the extremities.
Charcot-Marie-Tooth disease has a slow progression. Sometimes 10 years pass between manifestation and atrophy of the hands. Even if atrophy develops, patients can still work and care for themselves for a long time. With appropriate care, life expectancy remains at the level of the general population.
Classification of Charcot-Marie-Tooth disease
The genetic classification is very broad, with around 40 mutations affecting more than 20 different genes underlying the development of the disease. There are three main types of Charcot-Marie-Tooth disease based on morphological features and electromyographic data:
- Demyelinating type. It is characterized by destruction of the myelin sheath and the resulting reduction in impulse velocity (IPV) along the median nerve;
- Axonal type. The median nerve is characterized by normal or slightly reduced SPI because mainly axons are damaged;
- intermediate type. The impulse transmission speed is borderline.
This classification is useful for narrowing the search for a genetic diagnosis because some mutations have different clinical manifestations.
Effect
Severe deformities can only be corrected surgically because they make independent movement virtually impossible. However, surgical intervention is only indicated when conservative methods are ineffective.
The most common procedures are osteotomies – osteotomies of the heel bone or osteotomies of the metatarsals. In extreme cases, an arthrodesis is performed, in which the joint remains completely immobilized after the operation.
Arrange a consultation appointment online to avoid or delay surgery for Charcot disease for as long as possible. Our doctors will provide you with comprehensive information about any problems and symptoms that may arise. Our neurologists are there for you 24 hours a day.
frequently asked Questions
Charcot-Marie-Tooth syndrome is diagnosed more often in men than women.
The most important factor is the occurrence of Charcot-Marie-Tooth muscular atrophy in close family members. If both parents are carriers of the autosomal recessive gene, there is a 25 percent chance of having a child with this pathology. However, it may also be that the child is only a carrier of the gene, meaning that he or she will not show any symptoms over the course of his or her life: This risk is higher at 50 %. With X-linked inheritance, the risk of the gene being passed from mother to son is estimated at 50 %, but the child would only be a carrier. When a girl is born, the disease cannot be passed on, but her sons, that is, the grandchildren of patient zero, can inherit the defective gene. And they will have already developed Charcot-Marie-Tutte syndrome.
This mainly happens as a result of infections such as measles, rubella, infectious mononucleosis, pharyngitis or even a cold. Colds, head and spine injuries and vitamin deficiencies can also be contributing factors.
Clinical manifestations usually occur in children over 10 years old, less often between 16 and 30 years old.
classification
Clinical and genetic classification of hereditary motor neuropathies
Autosomal dominant hereditary motor-sensory neuropathy, type I:
– Caused by point mutations in the myelin gene PMR-22.
Type IB: Caused by point mutations in the Rho myelin gene.
Type IC: with an unidentified genetic defect.
Autosomal recessive hereditary motor-sensory neuropathy, type I: with unidentified genetic defect.
Autosomal dominant hereditary motor-sensory neuropathy, type II: with unidentified genetic defect.
Autosomal recessive hereditary motor-sensory neuropathy, type II: with unidentified genetic defect.
Hereditary motor-sensory neuropathy, type III:
– caused by point mutations in the myelin gene PMR-22;
– caused by point mutations in the myelin Rho gene.
X-linked sensory-motor neuropathy:
– caused by point mutations and deletions in connexin 32;
– with an unidentified genetic defect.
Complex forms of hereditary motor-sensory neuropathies:
– With optic nerve atrophy and deafness;
diagnosis
Diagnostic Criteria
Complaints and medical history: Muscle fatigue, muscle atrophy, 'polyneuric type' pain in the distal limbs, skeletal deformities, gait disorders, delay in motor development, progressive course.
History: Charcot-Marie disease: autosomal recessive inheritance, onset in the first decade of life.
In Degerin-Sott disease (hereditary motor-sensory neuropathy, type III): autosomal recessive inheritance, onset in the first two years of life, delayed motor development, weakness and atrophy of the distal limbs with progression to proximal lesions, ataxia, rapid course.
Hereditary motor-sensory neuropathy type I is characterized by an autosomal dominant inheritance type, with disease onset mainly in the 1st to 2nd.
Type II hereditary motor-sensory neuropathy is characterized by an autosomal dominant inheritance type, disease onset in the second decade of life, absent or only mild atrophy of the distal limbs, and a relatively mild course.
Hereditary motor-sensory neuropathy type II is characterized by an autosomal recessive type of inheritance, debut at a young age, pronounced atrophy and weakness of the distal limbs, deformities of the hands and feet, rapid progression of the course.
Physical examination: Neurological condition - pain (painful, shooting, cramping) in the distal limbs against the background of hypoesthesia, with gradual onset of weakness, atrophy in the distal limbs, reduction and loss of the Achilles reflex with progressive progression - carpo-radial; Gait disorder similar to 'gockeln', kicking.
Atrophy occurs in the distal limbs, with subsequent (progressive) atrophy in the proximal limbs. Deformed clubfoot or Friedreich's foot in the form of a hollow foot with a high arch and extension of the big toe phalanges. If the disease begins in childhood, there is a delay in motor development.
clinical picture
The first outbreak of the disease usually begins in adolescence. At first, the patient feels slight weakness in the lower limbs, which gradually increases.
Over time, fatigue increases with little physical exertion and muscle pain occurs. It becomes difficult to move due to the need to lift the legs, which is difficult to do due to the development of asymmetric atrophy. The legs, including the feet, may begin to go numb. Ten years after the first symptoms appear, patients are diagnosed with muscle weakness.
The symmetrical muscle weakness leads to the diagnosis of horse foot in all those affected. In the upper limbs, however, fewer than half of those affected experience the symptoms of Lou Gehrig's disease.
In Charcot disease, the main symptoms are reduced and complete loss of calf muscle contraction, patellar reflex and then ankle reflex. In addition, sensory disturbances occur in the upper and lower limbs and the affected person's gait changes.
'I have Marie Charco Tooth disease, how long will I live with this disease?
There is no exact information about life expectancy with amyotrophic sclerosis. Some patients can live their lives like normal people, others cannot. In primary amyotrophic lateral sclerosis, only the upper motor neuron is affected, and this form of Charcot disease allows patients to live long enough.
Progressive bulbar paralysis has the shortest life expectancy. A person with this form of pathology lives between six months and three years. Life expectancy with progressive muscular atrophy is on average five to 10 years.
diagnosis
The first step is to correctly distinguish the disease from progressive muscular atrophy, progressive bulbar paralysis, myopathy-nevinna and polyneuropathy with cryoglobulinemia.
Charcot disease often results in loss of speech. Therefore, as long as a person can still speak, it is worth teaching him to read lips.
Technology is moving with the times and almost every family has a computer, laptop or phone. Using special software, the patient can enter text into the device, which then reads it out loud.
Some families do not have this option or the patient is visually impaired. In this case, specially made cards can be used. Once it has been previously determined what the sign will mean, it is important to draw it clearly so that the patient can point to or show the card to express his or her wish.
In addition, you can try to guess what the patient wants yourself or use questions that only have a yes or no answer.
It should be noted that to date there is no effective treatment for Charcot disease. Basically, all therapies are aimed at stopping the further progression of the disease and eliminating the symptoms that have arisen as much as possible.
To date, there are a small number of drugs that prolong the life of a Charcot patient - one of them is Rilutec. This drug is essentially a blocker of tetrodotoxin (a powerful, naturally occurring non-protein poison) via sensitive sodium channels. Rilutec is only prescribed after consultation with your doctor, who should take a close look at your medical history. This is to ensure that there are no other diseases for which the drug is absolutely contraindicated.
The daily dose is 100 mg for life. The drug should be discontinued if you notice any abnormalities in the tests that are carried out regularly while taking Rilutec or its analogues. The drug is usually sold in tablet form.
Treatment approach
Treatment is tailored to the symptoms of Charcot Marie Tut Neuroma Amiotrophy. The interventions are complex and lifelong.
It should be noted that there are no more effective therapies in medicine. Only techniques that help alleviate the patient's symptoms and improve their quality of life are used.
It is important to optimize the patient's functional coordination and mobility. Therapeutic measures should be aimed at protecting weakened muscles from injury and reduced sensitivity.
The patient's relatives must support him in every way in the fight against the disease. After all, treatment takes place not only in medical institutions, but also at home.
All recommended measures must be carried out strictly and daily. Otherwise, the results of the treatment will not be achieved.
Treatment of amyotrophy involves a number of techniques:
- physiotherapeutic procedures;
- occupational therapy;
- a set of physical exercises;
- special foot support devices
- Orthopedic foot supports to correct a deformed foot;
- foot care;
- regular consultations with the attending physician;
- use of orthopedic surgery;
- injections of B vitamins;
- Administration of vitamins E, A and C.
- In amyotrophic lesions a special diet is prepared.. A diet rich in protein and potassium as well as an increased intake of vitamins is indicated.
- In the regressive course of the disease, mud bath treatment is carried out in parallel with the above measures. Recommended mud, rhodonium, igneous, sulfide and hydrogen sulfide baths. Electrophoresis treatment is also performed to stimulate the peripheral nerves.
- For joint misalignments and deformations of the skeletal system Correction by an orthopedist is indicated..
prevention
Prevention is. Consultation of a geneticist. Vaccinations against polio and tick-borne encephalitis should be given immediately.
Wearing comfortable orthopedic shoes can prevent the development of early foot deformities.
Patients should consult a podiatrist, that is, a foot specialist, who can prevent trophic soft tissue changes at an early stage and, if necessary, prescribe appropriate medication.
Walking difficulties can be caused by wearing special orthoses (Ankle-foot orthoses). These can control the flexion of the leg and lower leg on the back side, eliminate ankle instability and improve body balance.
Such a device allows the patient to move around without assistance and prevents unwanted falls and injuries. Foot splints are used for box foot syndrome.
Abroad there is a well-developed system of support for patients and their families: 'A world without Charcot disease without Marie Toutat'.
There are various specialized organizations, societies and foundations. Research is ongoing to find new treatments for this disease.
Unfortunately, there are no such institutions in the Russian Federation, but research to study and develop optimal treatment methods is ongoing and quite active.
Research institutes in Bashkortostan, Voronezh, Krasnoyarsk, Novokuznetsk, Samara, Saratov and Tomsk implement such programs.
Read more:- What is Charcot?.
- X-ray of Charcot's foot.
- What is distal?.
- Marfan Syndrome - Summary.
- paresis of the lower body.
- Shuffling Feet While Walking Causes.
- Syndrome of the tibial nerve.
- Innervation of the lower leg muscles.