Der Schweregrad der Erkrankung kann innerhalb einer Familie variieren. [2]

- Gummimann.
- Geschichte der Entdeckung des Syndroms
- Einführung
- Ursachen des Ehlers-Danlos-Syndroms
- Diagnose des Ehlers-Danlos-Syndroms
- Prognose beim Ehlers-Danlos-Syndrom
- Wie Ärzte das Ehlers-Danlos-Syndrom erkennen
- Wie das Ehlers-Danlos-Syndrom das Leben beeinflusst
- Symptome und Anzeichen des Ehlers-Danlos-Syndroms
- Komplikationen des Ehlers-Danlos-Syndroms
- Diagnose des Ehlers-Danlos-Syndroms
- Symptome und Anzeichen des Ehlers-Danlos-Syndroms
- Komplikationen des Ehlers-Danlos-Syndroms
- Diagnose des Ehlers-Danlos-Syndroms
- Das Ehlers-Danloh-Syndrom: ein wenig Geschichte
- Arten der Krankheit
- Bindegewebsdysplasie: ein aktuelles Thema
- Bindegewebsdysplasie: häufige klinische Syndrome
- Valvulares Syndrom
- Formen
- Diagnose des Ehlers-Danlos-Syndroms
Gummimann.

Das Ehlers-Danlos-Syndrom (einigen Quellen zufolge Ehlers-Danlos-Syndrom, EDS) ist eine seltene vererbte Kollagenpathologie, die die Haut, den Bewegungsapparat und andere Organe betrifft. Andere Bezeichnungen für dieses Syndrom sind Hyperelastizität der Haut, unvollkommene Desmogenese nach Rusakoff und Chernogubov-Ehlers-Danlos-Syndrom. Menschen mit diesem Syndrom wurden in der Vergangenheit in Zirkussen gesehen, weil sie mit ihrem Aussehen und ihren Bewegungen gewöhnliche Menschen überraschen konnten. Auch einige berühmte Persönlichkeiten hatten dieses Syndrom, wie der Geiger und Virtuose Niccolò Paganini.
Geschichte der Entdeckung des Syndroms
Im Jahr 400 v. Chr. schrieb Hippokrates in seinem Werk De aere, aquis, locis (Über Luft, Wasser und Orte) über Menschen mit dieser Krankheit. Er beobachtete Nomaden und Skythen mit schwachen Gelenken und zahlreichen Narben um sie herum. Hippokrates glaubte, dass es sich bei den Narben um Spuren einer Verätzung handelte, mit der man versuchte, die Hypermobilität der Gelenke zu verringern. Viel später – im Jahr 1657 – beschrieb der niederländische Chirurg Jansun van Meekeren (1611-1666) einen spanischen Jungen mit sehr elastischer Haut. Der Junge, der George Albes hieß, konnte die Haut an seinem Kinn bis zur Brust und die Haut an seinen Knien bis zur Mitte des Schienbeins dehnen, allerdings nur auf der rechten Körperseite. Der weltberühmte Geiger und Komponist Niccolò Paganini (1782-1840) hatte hypermobile Gelenke, einen schlanken Körperbau und eine deformierte Brust – alles Symptome, die typisch für einen Träger des Ehlers-Danlos-Syndroms sind. Gegen Ende des 19. Jahrhunderts traten einige SED-Patienten als Personen mit ungewöhnlichen Fähigkeiten in Wanderausstellungen auf – wie die von den amerikanischen Ärzten George M. Gould und Walter L. Pyle beschriebene ‚flexible lady‘. Die Vielfalt der klinischen Erscheinungsformen des Syndroms verhinderte lange Zeit eine detaillierte Beschreibung aller Formen der Krankheit.
Die erste ausführliche Beschreibung der SED wurde von Andrey Chernogubov, einem russischen Arzt des Moskauer Myasnitskaya-Krankenhauses, auf einer Tagung der Moskauer Gesellschaft für Venerologie und Dermatologie im Jahr 1892 vorgestellt. Er beschrieb zwei Patienten mit erhöhter Beweglichkeit der großen Gelenke. Bei dem einen handelte es sich um einen 17-jährigen Jungen mit Epilepsie, dessen Haut brüchig und hyperelastisch war und keine Nähte halten konnte.
Später, im Jahr 1901, veröffentlichte der dänische Dermatologe Edvard Lauritz Ehlers die Beschreibung eines Patienten mit schwachen Gelenken und hyperelastischer Haut sowie einer Neigung zu Blutergüssen. Auf einer Tagung der dänischen dermatologischen Gesellschaft im Jahr 1899 demonstrierte er diese Krankheit. Sieben Jahre später untersuchte der französische Arzt Henri-Alexandre Danlos einen Patienten mit vaskulären Hautveränderungen an den Ellenbogen und Knien. Einzelne Beschreibungen des Syndroms erschienen seitdem sowohl in den USA als auch in England, und bis 1966 stieg die Gesamtzahl der Berichte auf 300. 1936 brachte der englische Dermatologe Frederick Parkes Weber alle Fälle mit der Hyperelastizität und Brüchigkeit der Haut und der Hypermobilität der Gelenke in Verbindung. Er nannte die neue Erkrankung Ehlers-Danlos-Syndrom.
Einführung
Das Ehlers-Danlos-Syndrom (unvollkommene Desmogenese, hyperelastische Haut) gehört zusammen mit der Osteogenesis imperfecta, dem Marfan-Syndrom und anderen zu den erblichen Kollagenopathien. Das Ehlers-Danlos-Syndrom ist heterogen und umfasst eine heterogene Gruppe von erblichen Bindegewebsläsionen (Bindegewebsdysplasien), die mit einer gestörten Biosynthese von Kollagenproteinen einhergehen. Die Symptome des Ehlers-Danlos-Syndroms sind systemisch und betreffen u. a. das Muskel-Skelett-System, die Haut, das Herz-Kreislauf-System, das Augensystem und das Zahnsystem. Das Ehlers-Danlos-Syndrom ist daher nicht nur für die Genetik, sondern auch für die Traumatologie und Orthopädie, die Dermatologie, die Kardiologie, die Augenheilkunde und die Zahnmedizin von praktischem Interesse.
Die schwierige Verifizierung und das Vorhandensein leichter Formen erschweren es, genaue Informationen über die tatsächliche Häufigkeit des Ehlers-Danlos-Syndroms zu erhalten; die Diagnoserate der leichten Fälle liegt bei Neugeborenen bei 1:5 000, während die Häufigkeit der schweren Fälle bei 1:100 000 liegt.
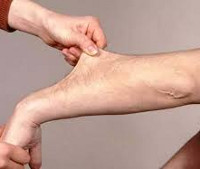
Ursachen des Ehlers-Danlos-Syndroms
Die verschiedenen Formen des Ehlers-Danlos-Syndroms unterscheiden sich durch die Art des Erbgangs und die primären molekularen und biochemischen Defekte. Alle klinischen Formen beruhen jedoch auf Genmutationen, die für quantitative oder strukturelle Anomalien des Kollagens verantwortlich sind. Bis heute sind die molekularen Mechanismen des Ehlers-Danlos-Syndroms nicht für alle Formen der Krankheit geklärt.
So ist beispielsweise bekannt, dass das Typ-I-Syndrom durch eine verminderte Fibroblastenaktivität, eine erhöhte Synthese von Proteoglykanen und einen Mangel an Enzymen, die für die normale Kollagenbiosynthese verantwortlich sind, gekennzeichnet ist. Das Ehlers-Danlos-Syndrom Typ IV geht mit einer unzureichenden Produktion von Kollagen des Typs III einher, und Typ VI ist durch einen Mangel an dem Enzym Lysylhydroxylase gekennzeichnet, das an der Hydroxylierung von Lysin in Prokollagenmolekülen beteiligt ist. Typ VII wird durch Anomalien bei der Umwandlung von Typ-I-Prokollagen in Kollagen verursacht; Typ X durch Anomalien des Plasmafibronektins, das an der Organisation der interzellulären Matrix beteiligt ist, usw.
Das pathomorphologische Bild bei den verschiedenen Typen des Ehlers-Danlos-Syndroms ist gekennzeichnet durch eine Verdünnung der Dermis, eine Verwirrung und einen Verlust der Kompaktheit der Kollagenfasern, eine Hypertrophie der elastischen Fasern, eine erhöhte Anzahl von Gefäßen und deren Lumen.
Diagnose des Ehlers-Danlos-Syndroms
Der Arzt stellt die Diagnose Ehlers-Danlos-Syndrom anhand der Symptome und der körperlichen Untersuchung. Zur Bestätigung der Diagnose werden in der Regel Gentests durchgeführt.
Der Arzt kann versuchen, bestimmte Arten des Ehlers-Danlos-Syndroms zu identifizieren, indem er eine Hautprobe zur Untersuchung unter dem Mikroskop entnimmt (Biopsie).
Andere Tests werden durchgeführt, um nach möglichen Krankheiten zu suchen, die mit Komplikationen verbunden sind. Zum Beispiel die Echokardiographie Echokardiographie und andere Ultraschallverfahren Bei einer Ultraschalluntersuchung werden Hochfrequenzwellen (Ultraschall) verwendet. Die von den inneren Strukturen reflektierten Wellen werden mit einem bewegten Bild aufgezeichnet. Röntgenstrahlen in. Lesen Sie weitere Informationen und andere bildgebende Verfahren zur Erkennung von Herz- oder Blutgefäßproblemen.
Prognose beim Ehlers-Danlos-Syndrom
Trotz der zahlreichen und vielfältigen Komplikationen ist die Lebenserwartung von Patienten mit Ehlers-Danlos-Syndrom im Allgemeinen normal. Bei einigen Menschen mit bestimmten Formen des Ehlers-Danlos-Syndroms sind die Komplikationen (in der Regel Blutungen) jedoch tödlich.
Es ist nicht möglich, das Ehlers-Danlos-Syndrom vollständig zu heilen oder Anomalien des Bindegewebes zu korrigieren. Verletzungen können behandelt werden, aber das kann für den Arzt schwierig sein, da beim Nähen der Schnitte das empfindliche Gewebe einreißen kann. In der Regel lassen sich Schnitte mit Klebeband oder medizinischem Hautkleber leichter schließen und hinterlassen weniger Narben.
Um Verletzungen zu vermeiden, sollten besondere Vorsichtsmaßnahmen getroffen werden. Kinder mit schwerem Ehlers-Danlos-Syndrom sollten zum Beispiel Schutzkleidung und Schutzschilde tragen.
Operationen erfordern spezielle Techniken, um Traumata zu minimieren und einen großen Vorrat an Blut für Transfusionen sicherzustellen. Der Geburtshelfer (ein auf die Geburt und die Betreuung und Behandlung der Schwangeren spezialisierter Arzt) sollte den Verlauf der Schwangerschaft und der Geburt überwachen. Eine genetische Beratung der Familienangehörigen wird empfohlen.
Copyright © 2023 Merck & Co, Inc, Rahway, NJ, USA und seine Tochtergesellschaften. Alle Rechte vorbehalten.
Wie Ärzte das Ehlers-Danlos-Syndrom erkennen
Die Cleveland Clinic, ein großes privates medizinisches Zentrum in den USA, erklärt, wie Ärzte EDS bei Patienten diagnostizieren:
- Gentests sind die häufigste Methode, um ein fehlerhaftes Gen zu identifizieren.
- Eine Biopsie ist ein Test, bei dem ein Arzt eine Hautprobe entnimmt und sie unter dem Mikroskop untersucht, um nach Krankheitsanzeichen wie bestimmten Genen und Genmutationen (Anomalien) zu suchen.
- Eine körperliche Untersuchung, bei der der Arzt feststellt, wie stark die Haut gedehnt ist und wie viel Bewegung die Gelenke haben.
- Röntgenaufnahmen und CT-Scans liefern Bilder von inneren Körperteilen, um Anomalien, Herzprobleme und Knochenverkrümmungen zu erkennen.
In Russland wird die SED-Diagnose auf der Grundlage einer visuellen Untersuchung und einer EMG-Elektromyografie gestellt, bei der der Zustand der Skelettmuskeln und -nerven beurteilt wird. Um die Art des SED-Syndroms zu bestimmen, können Tests auf Kollagenarten durchgeführt werden: Viele Privatkliniken bieten Tests zur Feststellung dieses Syndroms an.
Wie das Ehlers-Danlos-Syndrom das Leben beeinflusst
Das Ehlers-Danlos-Syndrom wirkt sich im Laufe des Lebens auf unterschiedliche Weise auf die Betroffenen aus. Es können andere Erkrankungen diagnostiziert werden: neurologische, Wirbelsäulen- oder Herz-Kreislauf-Erkrankungen. Bei der vaskulären Form des Syndroms kann es zum Beispiel zu Rissen in den Blutgefäßen kommen, die wiederum innere Blutungen oder Schlaganfälle verursachen können. Menschen mit diesem Typus haben auch ein erhöhtes Risiko, dass Organe reißen.
- Gelenk- und Muskelschmerzen, die mitunter sehr stark sein können;
- Schwächung des Bindegewebes, was zu Leistenbrüchen führen kann;
- schwächende chronische Müdigkeit, die sich negativ auf die geistigen und körperlichen Fähigkeiten auswirkt;
- Herz-Kreislauf-Störungen
- gastrointestinale Störungen;
- gynäkologische Probleme.
Darüber hinaus kann eine Person mit SED unter Schwindel, Gedächtnis- und Aufmerksamkeitsstörungen, verändertem Schwitzen und psychischen Problemen leiden.
Symptome und Anzeichen des Ehlers-Danlos-Syndroms
Die Symptome und Anzeichen des Ehlers-Danlos-Syndroms sind sehr unterschiedlich.
Die vorherrschenden Symptome sind Hypermobilität der Gelenke, abnorme Narbenbildung und gestörte Wundheilung, brüchige Blutgefäße und samtige, überdehnbare Haut. Die Haut kann um mehrere Zentimeter gedehnt werden, kehrt aber nach der Entlastung wieder in den Normalzustand zurück.
Breite, pergamentartige Narben bedecken oft knöcherne Vorsprünge, insbesondere an Ellenbogen, Knien und Unterschenkeln; beim hypermobilen Typ sind die Narben weniger ausgeprägt. Auf der Oberfläche von Narben oder Druckstellen bilden sich oft weiche Pseudotumore (fleischige Wucherungen).
Der Grad der Gelenkhypermobilität variiert, kann aber bei der Arthrochalasie, der klassischen und der hypermobilen Form erheblich sein.
Blutungsneigung ist selten, obwohl der vaskuläre Typ durch Gefäßrupturen und Blutergüsse gekennzeichnet ist. Subkutane verkalkte Kügelchen können tastbar oder auf Röntgenbildern sichtbar sein.
Komplikationen des Ehlers-Danlos-Syndroms
Kleinere Traumata können zu einer weit offenen Wunde führen, die jedoch nur wenig blutet; der chirurgische Verschluss von Wunden kann schwierig sein, da Nähte auf fragilem Gewebe zum Reißen neigen. Chirurgische Komplikationen entstehen aufgrund der Zerbrechlichkeit des tiefen Gewebes. Die Sklera kann sehr zerbrechlich sein, was zu kyphoskolioseartigen Augenperforationen führen kann.
Mäßige Synovialergüsse, Bandverstauchungen und Verrenkungen sind recht häufig. Vertebrale Kyphoskoliose entwickelt sich bei 25 % der Patienten (insbesondere bei Patienten mit Kyphoskoliose-Typ), Thoraxdeformitäten bei 20 % und Equinovarus-Klumpfuß Fußdeformität (Klumpfuß) und andere Fußanomalien Der Equinovarus-Klumpfuß ist gekennzeichnet durch Sohlenflexion, Einwärtsneigung der Ferse (von der Mittellinie des Fußes) und Vorfußadduktion (mediale Abweichung von der vertikalen Achse des Fußes). Lesen Sie mehr Informationen – unter 5%. Etwa 90 % der betroffenen Erwachsenen haben Plattfüße Der Equinovarus-Klumpfuß ist durch Plantarflexion, Einwärtsneigung der Ferse (von der Mittellinie des Fußes aus) und Vorfußadduktion (mediale Abweichung von der vertikalen Fußachse) gekennzeichnet. Lesen Sie mehr . Progressive Dysplasie des Hüftgelenks Progressive Dysplasie des Hüftgelenks (PDTS) Die progressive Dysplasie des Hüftgelenks (früher: kongenitale Hüftgelenkluxation) ist eine Entwicklungsanomalie des Hüftgelenks. (Siehe auch Einführung in kongenitale kraniofaziale und muskuloskelettale Dysplasien. Lesen Sie weitere Informationen (zuvor Kongenitale Hüftluxation) tritt in 1 % der Fälle auf (der arthrochalasische Typ ist durch eine bilaterale progressive Hüftdysplasie gekennzeichnet).
Diagnose des Ehlers-Danlos-Syndroms
Echokardiographie und/oder andere vaskuläre Untersuchungen zur Erkennung kardiovaskulärer Komplikationen
Die Erstdiagnose des Ellens-Danlos-Syndroms basiert hauptsächlich auf klinischen Befunden, muss jedoch durch Gentests bestätigt werden, mit denen sich inzwischen die meisten Subtypen identifizieren lassen. Die ultrastrukturelle Auswertung von Hautbiopsieproben kann helfen, die klassischen, hypermobilen und vaskulären Typen zu diagnostizieren.
Echokardiographie und andere bildgebende Verfahren für die Gefäße werden durchgeführt, um Herzerkrankungen (z. B. Klappenprolaps, Aneurysmen) zu erkennen, die bei einigen Typen auftreten können.
Symptome und Anzeichen des Ehlers-Danlos-Syndroms
Die Symptome und Anzeichen des Ehlers-Danlos-Syndroms sind sehr unterschiedlich.
Die vorherrschenden Symptome sind Hypermobilität der Gelenke, abnorme Narbenbildung und gestörte Wundheilung, fragile Blutgefäße und samtige, überdehnte Haut. Die Haut kann um mehrere Zentimeter gedehnt werden, kehrt aber nach der Entlastung wieder in den Normalzustand zurück.
Breite, pergamentartige Narben bedecken oft knöcherne Vorsprünge, insbesondere an Ellenbogen, Knien und Unterschenkeln; beim hypermobilen Typ sind die Narben weniger ausgeprägt. Auf der Oberfläche von Narben oder Druckstellen bilden sich oft weiche Pseudotumore (fleischige Wucherungen).
Der Grad der Gelenkhypermobilität variiert, kann aber bei der Arthrochalasie, der klassischen und der hypermobilen Form erheblich sein.
Die Neigung zu Blutungen ist selten, obwohl der vaskuläre Typ durch Gefäßrupturen und Blutergüsse gekennzeichnet ist. Subkutane verkalkte Kügelchen können tastbar oder auf Röntgenbildern sichtbar sein.
Komplikationen des Ehlers-Danlos-Syndroms
Ein kleines Trauma kann zu einer weit offenen Wunde führen, die jedoch nur wenig blutet; der chirurgische Wundverschluss kann schwierig sein, da Nähte auf fragilem Gewebe zum Reißen neigen. Chirurgische Komplikationen entstehen aufgrund der Zerbrechlichkeit des tiefen Gewebes. Die Sklera kann sehr zerbrechlich sein, was zu kyphoskolioseartigen Augenperforationen führen kann.
Mäßige Synovialergüsse, Bandverstauchungen und Verrenkungen sind recht häufig. Vertebrale Kyphoskoliose entwickelt sich bei 25 % der Patienten (insbesondere bei Patienten mit Kyphoskoliose-Typ), Thoraxdeformitäten bei 20 % und Equinovarus-Klumpfuß Fußdeformität (Klumpfuß) und andere Fußanomalien Der Equinovarus-Klumpfuß ist durch eine Beugung der Sohle, eine Einwärtsneigung der Ferse (von der Mittellinie des Fußes) und eine Vorfußadduktion (mediale Abweichung von der vertikalen Fußachse) gekennzeichnet. Lesen Sie weitere Informationen – auf Seite 5. Etwa 90 % der betroffenen Erwachsenen haben einen Plattfuß. Der Equinovarus-Klumpfuß ist gekennzeichnet durch Plantarflexion, Einwärtsneigung der Ferse (von der Mittellinie des Fußes aus) und Vorfußadduktion (mediale Abweichung von der vertikalen Fußachse). Lesen Sie mehr . Progressive Dysplasie des Hüftgelenks Progressive Dysplasie des Hüftgelenks (PDTS) Die progressive Dysplasie des Hüftgelenks (früher: kongenitale Hüftgelenkluxation) ist eine Entwicklungsanomalie des Hüftgelenks. (Siehe auch Einführung in kongenitale kraniofaziale und muskuloskelettale Dysplasien. Lesen Sie weitere Informationen (zuvor Kongenitale Hüftluxation) tritt in 1 % der Fälle auf (der arthrochalasische Typ ist durch eine bilaterale progressive Hüftdysplasie gekennzeichnet).
Diagnose des Ehlers-Danlos-Syndroms
Echokardiographie und/oder andere vaskuläre Untersuchungen zur Erkennung kardiovaskulärer Komplikationen
Die Erstdiagnose des Ellens-Danloh-Syndroms basiert hauptsächlich auf klinischen Befunden, muss jedoch durch Gentests bestätigt werden, mit denen inzwischen die meisten Subtypen identifiziert werden können. Die ultrastrukturelle Auswertung von Hautbiopsieproben kann helfen, die klassischen, hypermobilen und vaskulären Typen zu diagnostizieren.
Echokardiographie und andere bildgebende Verfahren für die Gefäße werden durchgeführt, um Herzerkrankungen (z. B. Klappenprolaps, Aneurysmen) zu erkennen, die bei einigen Typen auftreten können.
Das Ehlers-Danloh-Syndrom: ein wenig Geschichte
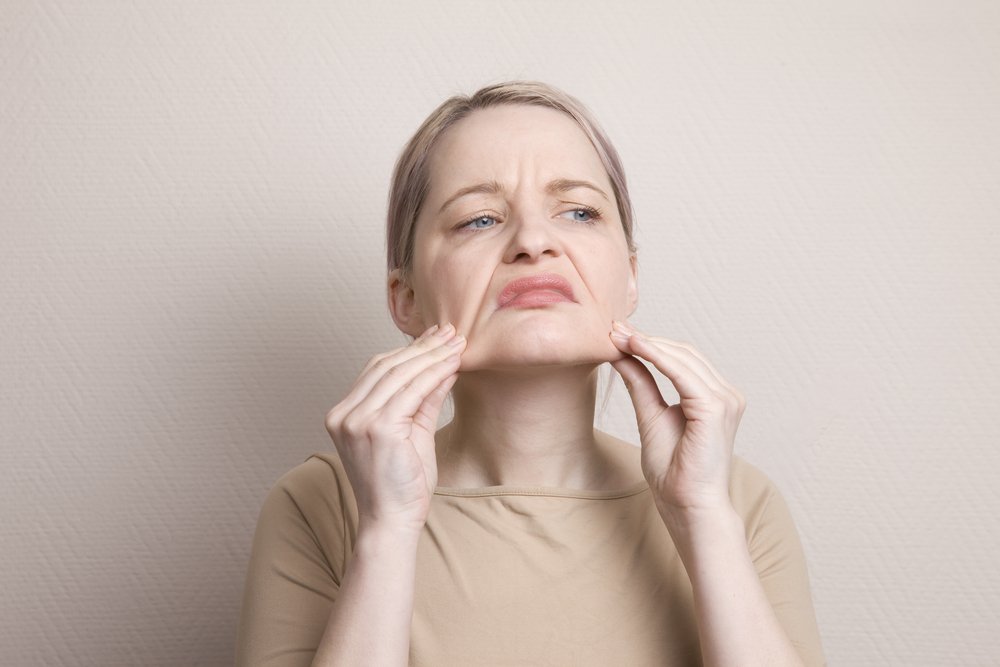
Die Krankheit wurde erstmals in den Schriften von Hippokrates erwähnt, der 400 v. Chr. Nomaden mit schwachen Gelenken beschrieb. Die Armen wurden durch Verätzung der Gelenke behandelt, um sie mit Verbrennungen zu vernarben und so die Hypermobilität der Gelenke zu minimieren.
Ein weiterer Arzt war ein niederländischer Chirurg, J. J. K. Van Meekeren, der einen spanischen Jungen mit unglaublich elastischer Haut beschrieb. Das Kind konnte seine Haut von den Knien bis zur halben Höhe der Schienbeine dehnen, und die Haut von seinem Kinn reichte leicht bis zur halben Höhe der Brust. Es stimmt, dass nur die Haut auf der rechten Körperseite des Jungen eine so bemerkenswerte Elastizität aufwies.
Die erste detaillierte Beschreibung der Krankheit stammt von Andrej Tschernogubow, einem russischen Dermatologen, der Ende des 19. Jahrhunderts im Moskauer Mjasnizkaja-Krankenhaus arbeitete. Der Arzt stellte 1892 auf einer Tagung der Moskauer Gesellschaft für Venerologie und Dermatologie Beschreibungen von zwei Patienten vor. Im Jahr 1901 veröffentlichte der Däne Edward Ehlers die Beschreibung eines Patienten mit hyperelastischer Haut, schwachen Gelenken und einer starken Neigung zu Blutergüssen. Sieben Jahre später wurde ein weiterer Patient von dem französischen Dermatologen Henri Danleau beschrieben. Bis 1966 stieg die Zahl der beschriebenen Fälle auf 300. Doch bereits 1936 hatte der englische Dermatologe Frederick Weber die vorhandenen Beschreibungen systematisiert und Fälle von Gelenkhypermobilität, Hautbrüchigkeit und Hyperelastizität zu einem einzigen Krankheitsbild zusammengefasst, das er Ehlers-Danlos-Syndrom nannte. Offensichtlich kannte Weber das Tschernogubow-Syndrom nicht oder ignorierte dessen Vorrangstellung. Heute gibt es mehrere Varianten des Namens der Krankheit. Der Kürze halber werden wir uns im weiteren Verlauf dieses Artikels auf die gebräuchlichste Abkürzung beziehen – SED. Wir sollten jedoch nicht vergessen, dass der erste in der neuen Geschichte immer noch Tschernogubow war.
Arten der Krankheit
SED ist eine Erbkrankheit, bei der die Molekularstruktur des Kollagens gestört ist. Sie beeinträchtigt mehr als nur die Eigenschaften der Haut. Ende des 20. Jahrhunderts hatten die Wissenschaftler bereits sechs Haupttypen der Krankheit identifiziert:
- klassisch;
- hypermobile
- vaskulär;
- Kyphoskoliose;
- Achondroplasie;
- Dermatosparaxis.
Alle sind mit einem Defekt in Genen verbunden, die die Kollagensynthese beeinflussen, aber sie treten auf unterschiedliche Weise und mit unterschiedlicher Häufigkeit auf. ‚Fehlerhafte‘ Gene führen dazu, dass die Kollagenfasern ihre charakteristische Struktur verlieren. Mutationen können zufällig auftreten, aber es gibt auch bekannte Fälle von familiärer SED. Insbesondere in dem aserbaidschanischen Dorf Gobu im Bezirk Apsheron ist die Geburtenrate von Kindern mit SED mit etwa 10 % viel höher als in anderen Gebieten.
- Die klassische SED ist nicht die häufigste Form der Krankheit – auf 20-40.000 Menschen kommt ein Patient mit hyperextrahierter Haut, zerbrechlichen Blutgefäßen, die leicht zu Blutergüssen führen, und Wunden, die unter Bildung atypischer Narben und Narbenbildung heilen. Subkutane Zysten und gutartige Tumore sind ebenfalls häufig.
- Die hypermobile EDM ist häufiger: ein Fall pro 10 000 bis 15 000 Menschen. Das Hauptsymptom ist die Schwäche der kleinen und großen Gelenke, die leichte Verrenkbarkeit und die frühe Entwicklung von Osteoporose. Gastrointestinale Störungen wie Darmbeschwerden, gastroösophagealer Reflux und Gastritis sind ebenfalls charakteristisch für diese Form.
- Die vaskuläre Form der SED ist seltener: ein Fall pro 250 000 Menschen. Aufgrund der Brüchigkeit der Blutgefäße kommt es häufig zu schweren Blutungen, sowohl äußerlich als auch innerlich. Aus diesem Grund übersteigt die Lebenserwartung dieser Patienten selten 50 Jahre. Bei dieser Form liegt keine ausgeprägte Hyperelastizität der Haut vor.
- Der achondroplasmatische Typ ist noch seltener: nur 30 Fälle sind beschrieben worden. Er ist gekennzeichnet durch eine angeborene Hüftluxation, eine Neigung zu Blutergüssen und Narben, eine Veranlagung zu früher Arthritis und eine sehr elastische Haut.
Bindegewebsdysplasie: ein aktuelles Thema
Der Begriff ‚Dysplasie‘ bezieht sich auf eine Störung bei der Bildung einer bestimmten Struktur. In unserem Fall ist es das Bindegewebe. Es handelt sich um einen angeborenen Zustand, bei dem Mutationen in den Genen vorliegen, die für die Entwicklung von Bindegewebskomponenten verantwortlich sind.
Diese Pathologie ist weit verbreitet. So wird in Russland laut Statistik bei einem von 10 Einwohnern eine Bindegewebsdysplasie diagnostiziert. Verschiedenen Quellen zufolge haben 10-70 % der Weltbevölkerung diese Diagnose.
Diese Pathologie ist schwer zu diagnostizieren – über viele Jahre hinweg kann der Patient Spezialisten verschiedener Fachrichtungen mit unterschiedlichen Beschwerden konsultieren, ohne eine eindeutige Diagnose zu erhalten.
Bindegewebsdysplasie: häufige klinische Syndrome

Die Bindegewebsdysplasie ist eine Krankheit mit einer Vielzahl von klinischen Erscheinungsformen, deren Schweregrad von mehreren Faktoren abhängt:
- der Art und Anzahl der Mutationen
- der Art des betroffenen Gewebes (lockeres oder dichtes Bindegewebe);
- der Art der Anomalie in der Phase der Bindegewebsbildung.
Da Bindegewebe ein integraler Bestandteil vieler Organe und Systeme ist, ist die Krankheit multisystemisch. Bei jedem Patienten kann sie einen anderen Verlauf nehmen und mehr oder weniger spezifische Organe betreffen. Daraus ergibt sich ein vielfältiges klinisches Bild.
In der Medizin wird üblicherweise zwischen den Formen der Bindegewebsdysplasie unterschieden:
Die schwersten Symptome, die in ständiger Kombination auftreten, werden in verschiedene Syndrome eingeteilt. Es handelt sich um verschiedene Formen von Dysplasie:
Diese Syndrome sind recht selten, das klinische Bild dieser Krankheiten ist auffällig und die Diagnose ist in der Regel unzweifelhaft.
Problematisch ist die undifferenzierte Dysplasie, die nicht in das klinische Bild eines der differenzierten Syndrome passt. Das Vorhandensein dieser Merkmale, die kein eindeutiges klinisches Bild ergeben, aber für die Patienten besorgniserregend sind und ihre Lebensqualität beeinträchtigen, wird als undifferenzierte Bindegewebsdysplasie bezeichnet.
Das Interessante daran ist, dass jeder Patient einzigartig ist. Es gibt sozusagen keine ‚Standard‘-Symptome und -Zeichen für diese Krankheit. Daher werden in der Regel mehrere Syndrome diagnostiziert, die auf der Dominanz des einen oder anderen Systems beruhen.
Valvulares Syndrom
Bereits im Kindesalter werden bei ‚dysplastischen Kindern‘ Veränderungen im Herzklappenapparat diagnostiziert. Die häufigste Diagnose ist der Mitralklappenprolaps. Läsionen können auch die Aorten- und Trikuspidalklappen betreffen, sind aber seltener.
Formen
Die Ehlers-Danlos-Syndrome umfassen eine heterogene Gruppe von Erkrankungen, die durch eine Fragilität des weichen Bindegewebes und weit verbreitete Manifestationen in der Haut, den Bändern und Gelenken, den Blutgefäßen und den inneren Organen gekennzeichnet sind. Das klinische Spektrum reicht von leichter Hyperelastizität der Haut und Gelenke bis hin zu schweren körperlichen Behinderungen und lebensbedrohlichen Gefäßkomplikationen.
Ursprünglich wurden die 11 Formen des Ehlers-Danlos-Syndroms mit römischen Ziffern benannt (Typ I, Typ II, usw.). Im Jahr 1997 schlugen Forscher eine einfachere Klassifizierung (Villefranche-Nomenklatur) vor, die die Anzahl der Typen auf sechs reduzierte und ihnen beschreibende Namen auf der Grundlage ihrer Hauptmerkmale gab. [7]
Die aktuelle Villefranche-Klassifikation kennt sechs Subtypen, von denen die meisten mit Mutationen in einem der Gene verbunden sind, die für kollagene Fibrillenproteine oder Enzyme kodieren, die an der posttranslationalen Modifikation dieser Proteine beteiligt sind. [8]
- Klassischer Typ I (OMIM 606408)
- Typ II Klassischer Typ, Ehlers-Danlos-Syndrom mit Tenascin-X-Mangel
- Typ III Hypermobilitätstyp
- Typ VIA, Typ VIB Vaskulärer Typ (OMIM 225320)
- Typ VIIA und VIIB Arthrochalasie-Typ (OMIM 130060, 617821), Typ VIIC Dermatosparaxis (OMIM 225410), Progeroid-Typ
- Typ VIII Parodontitis-Typ, Ehlers-Danlos-Variante mit periacetabularer Heterotopie
Die Bestimmung des richtigen EDS-Subtyps hat wichtige Auswirkungen auf die genetische Beratung und Behandlung und wird durch spezifische biochemische und molekulare Tests begünstigt. [9]
Diagnose des Ehlers-Danlos-Syndroms
Der Umfang der Untersuchung hängt vom Vorhandensein der wichtigsten klinischen Merkmale der Krankheit ab. Genealogische Studien und molekulargenetische Methoden sind erforderlich.
Die folgenden Voraussetzungen sind für die Diagnose des Ehlers-Danloh-Syndroms erforderlich.
- Das Vorhandensein von mindestens einem Hauptkriterium ist für die klinische Diagnose erforderlich. Bei ausreichender Kapazität garantiert das Vorhandensein eines oder mehrerer Hauptkriterien die Bestätigung des Ehlers-Danloh-Syndroms auf Laborebene.
- Ein Nebenkriterium ist ein Merkmal, das einen geringeren Grad an diagnostischer Spezifität aufweist. Das Vorhandensein eines oder mehrerer Nebenkriterien trägt zur Diagnose eines bestimmten Typs des Ehlers-Danlos-Syndroms bei.
- Liegen keine großen Kriterien vor, reichen kleine Kriterien nicht aus, um die Diagnose zu stellen. Das Vorhandensein kleiner Kriterien deutet auf das Vorhandensein eines dem Ehlers-Danlos-Syndrom ähnlichen Zustands hin, dessen Natur klarer wird, wenn seine molekulare Grundlage bekannt wird. Da die Prävalenz der kleinen Kriterien viel höher ist als die der großen Kriterien, bildet in voller Übereinstimmung mit der Revision von Wilfranche das Vorhandensein von nur kleinen Kriterien die Grundlage für die Diagnose eines Ehlers-ähnlichen Phänotyps.
Die Diagnose des klassischen Syndroms wird bei einem Patienten auf der Grundlage der minimalen klinischen Diagnosekriterien (Hyperelastizität der Haut und Vorhandensein atrophischer Narben) und der Identifizierung des pathogenen COL5A1-, COL5A2- oder COL1A1-Gens durch molekulargenetische Tests gestellt.
Zu den diagnostischen Kriterien für das Morfan-Syndrom und das Ehlers-Danloh-Syndrom gehört auch eine Hypermobilität der Gelenke. Wenn die entsprechenden Kriterien nicht erfüllt sind, sollte die Hypermobilität als eigenständige Erkrankung behandelt werden.
Lesen Sie mehr:- Fußwurzel- und Mittelfußgelenke.
- Marfan-Syndrom bei Neugeborenen.
- Das Marfan-Syndrom auf einen Blick.
- Marfan-Syndrom.
- Marfan-Syndrom – Zusammenfassung.
- Behandlung des Kurzbeinsyndroms.
- Streckung und Beugung des Fußes.
- Fußwurzel.