Zu den Medikamenten, die zur Behandlung von Symptomen eingesetzt werden, die auf eine Herz-Kreislauf-Erkrankung hindeuten, gehören Betablocker, Angiotensin-konvertierende Enzyminhibitoren und Kalziumantagonisten. Eine Herzklappeninsuffizienz, ein thorakales Aortenaneurysma mit einem Durchmesser von fünf oder mehr Zentimetern und eine Aortendissektion sind Indikationen für einen chirurgischen Eingriff.

- Marfan-Syndrom
- Pflege auf höchstem Niveau: Was moderne Windeln leisten können
- Symptome
- Ursachen des Marfan-Syndroms
- Wie wird diagnostiziert?
- Ursachen des Marfan-Syndroms
- Klassifizierung des Marfan-Syndroms
- Symptome des Marfan-Syndroms
- Muskuloskelettales System
- Warum sollte ein Arzt konsultiert werden, wenn Symptome des Marfan-Syndroms festgestellt werden?
- Das Marfan-Syndrom – eine Krankheit der Genies?
- Behandlung des Marfan-Syndroms
- Was sollten Ärzte über das Marfan-Syndrom wissen?
- Symptome aus verschiedenen Organen und Systemen
- Hauptanzeichen der Pathologie
- Wie kann man eine Krankheit bei einem Kind genau diagnostizieren?
- Diagnose des Marfan-Syndroms
- Prognose beim Marfan-Syndrom
- Was sind die Symptome des Marfan-Syndroms?
- Wie erfolgt die Behandlung des Marfan-Syndroms?
- Wie äußert sich das Marfan-Syndrom?
- Behandlungsgrundsätze für das Marfan-Syndrom
Marfan-Syndrom
Das Marfan-Syndrom ist eine systemische, autosomal-dominant vererbte Krankheit, die durch eine Unterentwicklung der Bindegewebsfasern aufgrund von Strukturdefekten im Kollagen gekennzeichnet ist. Die am häufigsten betroffenen Organe sind die Augen, der Bewegungsapparat, das Herz und die Blutgefäße. Die Prognose der Krankheit hängt von der Schwere der begleitenden pathologischen Veränderungen ab. Die meisten Patienten werden nicht älter als 50 Jahre.
Die charakteristischen Symptome des Marfan-Syndroms wurden erstmals 1875 von amerikanischen Ärzten beschrieben. Benannt ist die Krankheit nach dem französischen Kinderarzt A. Marfan, der die Krankheit mehrere Jahre lang erforschte.
Heute ist das Marfan-Syndrom nicht sehr häufig. Die durchschnittliche Inzidenz liegt zwischen 1:20 000 und 1:5 000, wobei Männer und Frauen gleich häufig betroffen sind. Es gibt auch kein rassisches Muster.

Pflege auf höchstem Niveau: Was moderne Windeln leisten können
Symptome
Die Symptome des Marfan-Syndroms sind vielfältig.
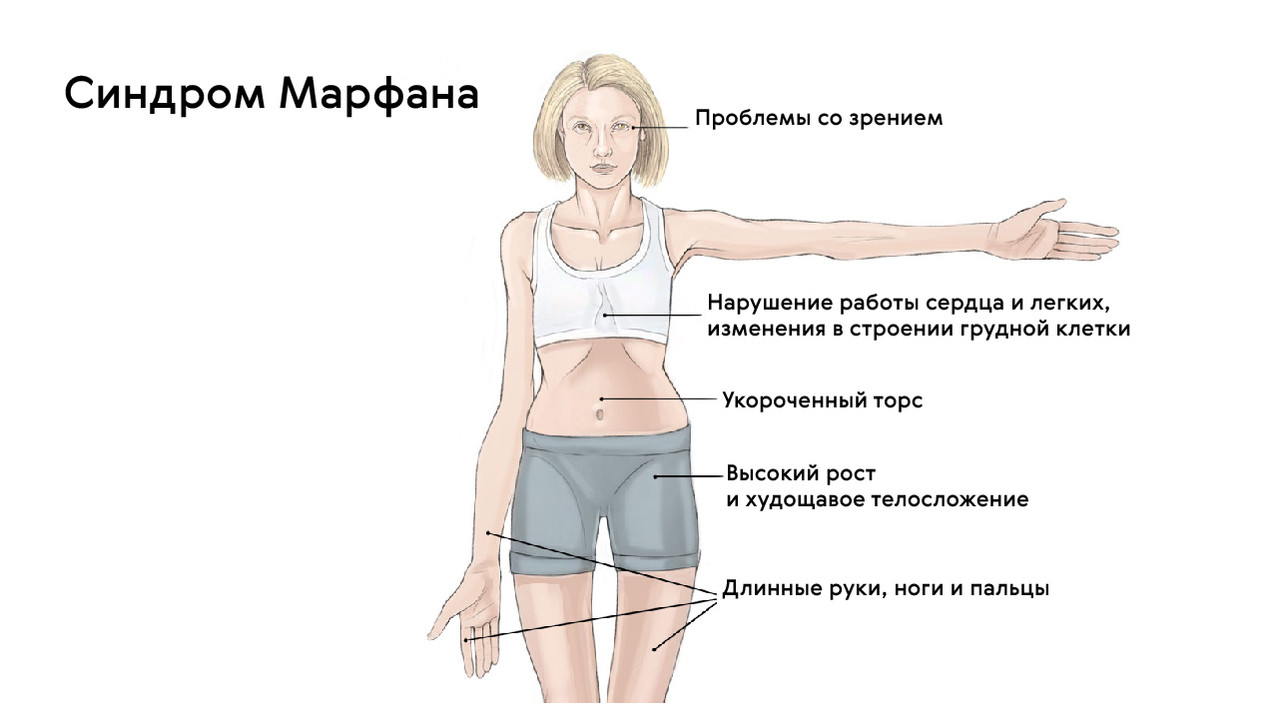
Patienten mit dieser Krankheit haben ein auffälliges Aussehen. Sie sind groß, haben einen relativ kurzen Rumpf und übermäßig verlängerte obere und untere Gliedmaßen. Die Arme und Beine der Patienten sind nicht nur unverhältnismäßig lang, sondern auch sehr dünn. Die Finger sind gestreckt und dünn, das Unterhautfettgewebe ist unterentwickelt und der Muskeltonus ist reduziert.
Der Kopf des Patienten ist lang und schmal. Nicht selten liegt eine Anomalie im Oberkiefer vor, bei der sich der harte Gaumen nach oben wölbt und einen hohen Bogen bildet. Der unregelmäßige Biss ist das Ergebnis von Kieferfehlstellungen.
Die Gelenke des Patienten sind sehr beweglich (hypermobil), und der Brustkorb ist deformiert, wobei das Brustbein nach vorne ragt (Kielform) oder nach innen fällt (Trichterform). Hinzu kommen verschiedene Wirbelsäulendeformitäten wie die Krümmung in der anteroposterioren Ebene.
Typisch für das Marfan-Syndrom sind verschiedene Anomalien des Herz-Kreislauf-Systems. Diese können sich in Form von Gefäßwanddefekten wie Aneurysmen, Klappen- und Septumdefekten wie Mitralklappenprolaps, Aortenwurzelerweiterung und so weiter äußern. Im Jahr 2013 veröffentlichten Forscher des Nationalen Medizinischen Forschungszentrums für Herz- und Gefäßchirurgie A.N. Bakulev eine Reihe von Studien über die Entstehung von Herzaneurysmen. Das Nationale Medizinische Forschungszentrum Bakoulev veröffentlichte eine Studie, aus der hervorging, dass bei 60 % der Patienten mit Marfan-Syndrom eine Aortenwurzelerweiterung und bei 91 % ein Mitralklappenprolaps auftritt.
Darüber hinaus sind Sehstörungen eine häufige Folge der Krankheit. Klinisch kann sich dies als Myopie unterschiedlichen Schweregrades, Linsenektopie, Strabismus, Hornhautanomalien usw. äußern.
Ursachen des Marfan-Syndroms
Es tritt bei 1 von 10 000 Menschen auf. Der Entstehung des Marfan-Syndroms liegt ein Gendefekt auf Chromosom 15 zugrunde, der die Struktur des Fibrillins bestimmt. Dieses Protein ist der Hauptbestandteil der Elastin-assoziierten Mikrofibrillen. Letztere finden sich in den meisten Bindegeweben, großen Blutgefäßen und Bändern, die die Linse stützen. Die Krankheit wird autosomal-dominant vererbt: Wenn ein Elternteil die Krankheit hat, besteht eine 50 %ige Chance, dass das Kind die Pathologie entwickelt. Es gibt Fälle, in denen ein gesundes Paar ein Kind mit dieser genetischen Anomalie bekommt: eine spontane Mutation tritt in 1 von 4 Fällen auf.
- kielförmige oder trichterförmige Krümmung des Brustkorbs, Wirbelsäulendeformität
- überproportionaler Rumpf, hohe Statur
- lange (gekrümmte) Finger (wenn das Handgelenk positiv ist, überlappen sich Daumen und Zeigefinger, wenn die andere Hand gefesselt ist)
- Vergrößerung des Bewegungsumfangs der Gliedmaßen
- abnorme Gelenkbeweglichkeit
- flache Valgusdeformität des Fußes
- Gewichtsmangel
- gewölbter Gaumen, Deformierung des Gebisses
- atypisches Gesicht (Dolichocephalie, Unterentwicklung des Jochbeins, eingefallene Augen)
- Sehstörungen durch eine ektopische Linse, abnorme Hornhautabflachung, Kurzsichtigkeit usw.
- Dyspnoe, Schwäche
- Hautstriae
- Wiederkehrende Leistenbrüche, etc.
Wie wird diagnostiziert?
Der diagnostische Behandlungsalgorithmus hängt von den betroffenen Systemen ab. Die Konsultation folgender Fachärzte ist erforderlich: Genetiker, Augenarzt, Kardiologe, Pulmonologe, Gastroenterologe.
Die instrumentelle Untersuchung stützt sich auf:
- Röntgenaufnahmen oder Computerbilder von Händen, Füßen, Brustkorb, Wirbelsäule, Hüftknochen und Schädel.
- Magnetresonanztomographie (MRT) – wenn wir Veränderungen der inneren Organe diagnostizieren wollen: Anomalien des Darms, Herzfehler, Nieren, usw. Die thorakoabdominale Magnetresonanzangiographie ermöglicht eine vollständige Beurteilung des Arteriensystems.
- EKG, Ultraschall, transthorakale Echokardiographie (Echokardiographie) des Herzens und der Gefäße;
Ursachen des Marfan-Syndroms
Das Marfan-Syndrom ist eine autosomal dominante angeborene Anomalie mit ausgeprägtem Pleiotropismus, variabler Ausprägung und hoher Penetranz. Dem Marfan-Syndrom liegen Mutationen im FBN1-Gen zugrunde, das für die Synthese von Fibrillen verantwortlich ist, einem wesentlichen Strukturprotein der interzellulären Matrix, das dem Bindegewebe Elastizität und Kontraktilität verleiht. Die Anomalie und der Mangel an Fibrillin beim Marfan-Syndrom führen zu einer gestörten Bildung faseriger Strukturen, einem Verlust der Festigkeit und Elastizität des Bindegewebes und der Unfähigkeit, physiologischen Belastungen standzuhalten. Die elastischen Gefäßwände und der Bandapparat (insbesondere die Aorta und das Zimtband des Auges, die den höchsten Anteil an Fibrillin enthalten) sind anfälliger für histologische Veränderungen.
Das breite phänotypische Spektrum des Marfan-Syndroms (von milden, schwer zu unterscheidenden bis hin zu schweren, rasch fortschreitenden Formen) ist auf die Vielfalt der Mutationen im FBN1-Gen (mehr als 1.000 Typen) sowie auf das Vorhandensein von Mutationen in anderen Genen (z. B. dem transformierenden Wachstumsfaktor-Gen TGFBR-2) zurückzuführen. Gentests zeigen in 75 Prozent der Fälle des Marfan-Syndroms einen familiären Vererbungstyp und in den übrigen Fällen eine primäre Mutation. Das Risiko, ein Kind mit Marfan-Syndrom zu bekommen, steigt mit dem Alter des Vaters (insbesondere nach dem 35. Lebensjahr).
Klassifizierung des Marfan-Syndroms
Je nach Anzahl der betroffenen Systeme gibt es verschiedene Formen des Marfan-Syndroms
- Steril – mit leichten Veränderungen in 1 oder 2 Systemen
- schwer – mit leichten Läsionen in 3 Systemen; schwere Läsionen in mindestens 1 System; schwere Läsionen in 2-3 oder mehr Systemen.
Der Schweregrad der Veränderungen beim Marfan-Syndrom kann leicht, mittelschwer oder schwer sein. Es wird zwischen einem progressiven und einem stabilen Marfan-Syndrom unterschieden.
Symptome des Marfan-Syndroms
Die Symptome des Gendefekts können von geringfügigen Veränderungen des Bindegewebes bis hin zu schweren Störungen wesentlicher Körperfunktionen reichen. Mit anderen Worten: Die äußeren Erscheinungsformen der Anomalie können trotz des gleichen Gendefekts von Patient zu Patient sehr unterschiedlich sein.
Die klassische Trias des Marfan-Syndroms besteht aus Skelettanomalien, Linsenverschiebung und Aortendissektion (Abbildung 2). Außerdem verursachen systemische Bindegewebsschäden bei Patienten Anomalien in fast allen Organen und Systemen des Körpers.
Muskuloskelettales System
Der Schweregrad der Muskel-Skelett-Symptome hängt von der Schwere des Falls und den Körpereigenschaften des Patienten ab.
Menschen mit Marfan-Syndrom sind außergewöhnlich groß: Die Kinder neigen dazu, alle Familienmitglieder zu überragen. Häufig wird eine abnorme Armlänge festgestellt, insbesondere bei Kindern, bei denen die Arme länger als der Körper sind.
Ein auffälliges Symptom sind abnorm verlängerte und dünne Finger, sogenannte Spinnenfinger (Arachnodaktylie) (Abbildung 3).
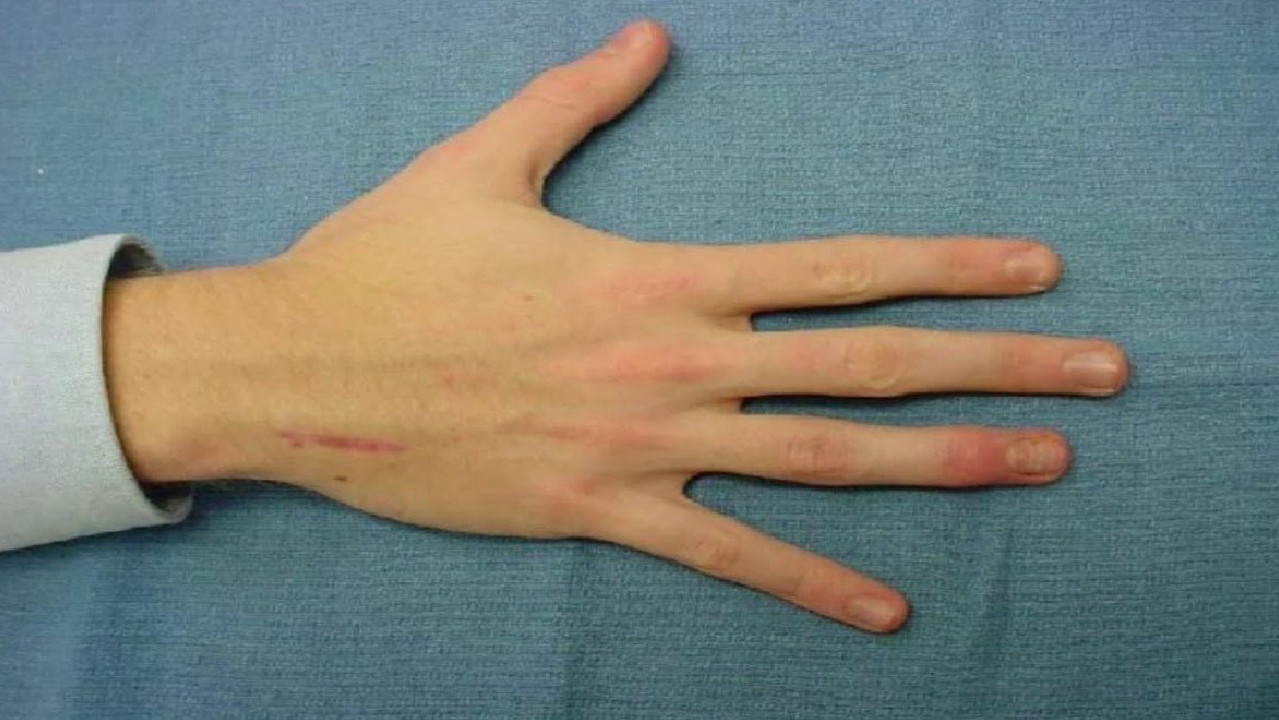
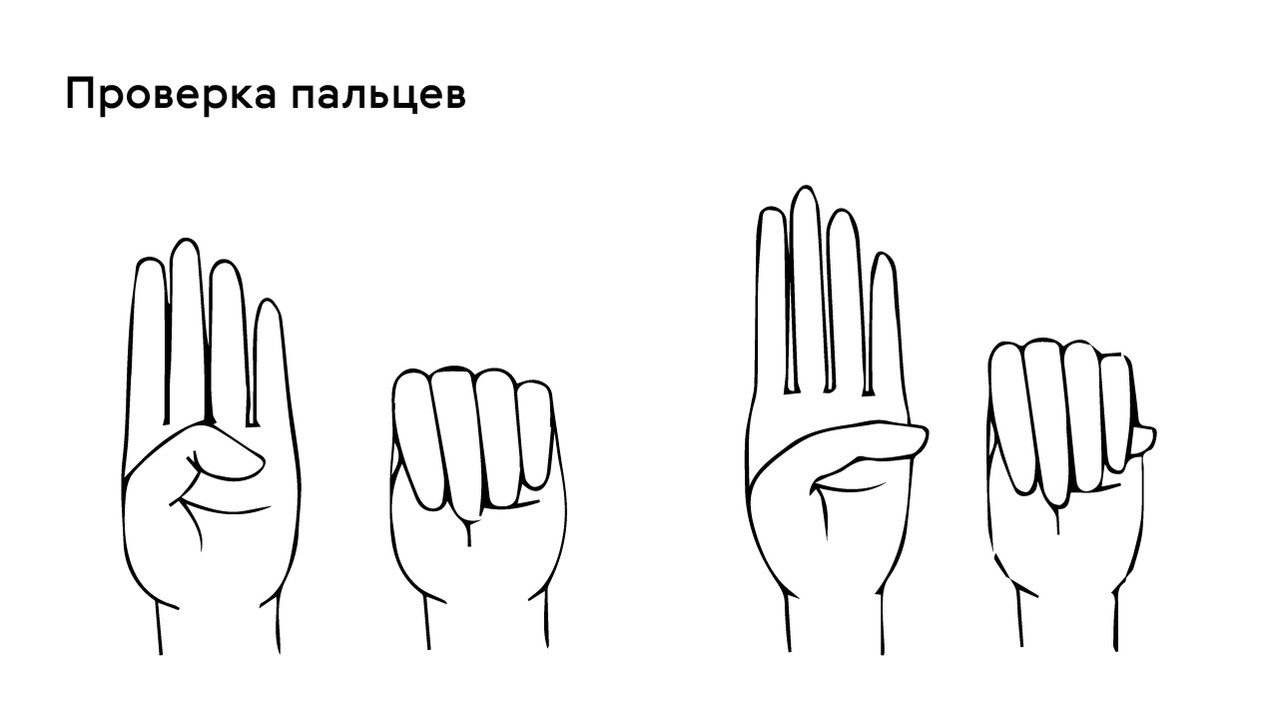
Das Vorhandensein dieses Symptoms kann mit dem Daumentest überprüft werden: Bei Patienten mit Arachnodaktylie ragt ein Teil des Daumens (distales Fingerglied) über den Rand der geballten Faust hinaus (Abb. 4).
Das Gesicht von Menschen mit Marfan-Syndrom ist meist länglich und dünn. Dies ist auf die hohe Position des oberen Gaumens, den verlängerten Schädel und die pathologische Schlankheit zurückzuführen.
Sie sind auch durch Thoraxdeformitäten gekennzeichnet, die in zwei Varianten auftreten können: nach innen (trichterförmiger Brustkorb) oder nach außen (kielförmiger Brustkorb, Abbildung 5).
Warum sollte ein Arzt konsultiert werden, wenn Symptome des Marfan-Syndroms festgestellt werden?
Die genetische Anomalie selbst ist mit dem Leben vereinbar. Die Folgen der durch die FBN1-Mutation verursachten Krankheit sind jedoch gefährlich:
- Ruptur der großen Blutgefäße, am häufigsten der Aorta;
- Chronische Herzinsuffizienz – die Unfähigkeit des Herzens, alle Organe mit dem notwendigen Blut zu versorgen;
- Verminderte Sehschärfe oder vollständiger Verlust der Sehfunktion.
Die Ruptur eines Aneurysmas der Aorta oder eines anderen großen Gefäßes führt oft zum sofortigen Tod. Die chronische Herzinsuffizienz kann in eine akute Form übergehen und ohne dringende medizinische Behandlung auch tödliche Folgen haben – den plötzlichen Koronartod. Es sind diese Komplikationen, die bei Kindern mit Marfan-Syndrom am häufigsten zum Tod führen. Eine Frau mit FBN1-Genmutationssyndrom ist während der Schwangerschaft besonders gefährdet: Die erhöhte Belastung der Aorta vervielfacht das Risiko einer Aortenruptur.
Um gefährlichen Komplikationen vorzubeugen und die daraus resultierenden Störungen zu kompensieren, sollten Eltern beim ersten Verdacht auf ein Marfan-Syndrom bei ihrem Kind so schnell wie möglich ärztliche Hilfe in Anspruch nehmen. Es ist wichtig, das Kind nicht nur einmal zu untersuchen, sondern sich auch bei den Ärzten anzumelden, die an der Behebung der Symptome beteiligt sind:
- einem Spezialisten für Genetik;
- einem Kardiologen
- einem Spezialisten für Wirbelsäulenorthopädie;
- einem Dermatologen
- Augenarzt;
- Gastroenterologe.
Die Liste der Spezialisten hängt von der Schwere der Erkrankung ab, und regelmäßige umfassende Vorsorgeuntersuchungen ermöglichen die frühzeitige Erkennung neuer Anomalien.
Das Marfan-Syndrom – eine Krankheit der Genies?
Das Marfan-Syndrom ist nicht nur mit vielen Gründen für Arztbesuche verbunden. Menschen mit einer Mutation im FBN1-Gen kompensieren die körperlichen Symptome der Krankheit oft mit intellektuellen Fähigkeiten, weshalb diese genetische Störung auch als ‚Genie-Syndrom‘ bezeichnet wird. Es wird vermutet, dass die erhöhte Adrenalinausschüttung als Folge pathologischer Veränderungen in den Nebennieren für die hohe geistige und psychische Aktivität dieser Patienten verantwortlich ist. Daher finden sich unter den Menschen mit Marfan-Syndrom auch berühmte Persönlichkeiten. Julius Caesar, Abraham Lincoln und Charles de Gaulle zum Beispiel wurden durch die Pathologie nicht daran gehindert, berühmte Politiker zu werden; Hans Christian Andersen und Korney Chukovsky schufen außergewöhnliche literarische Werke und Niccolò Paganini wurde als brillanter Musiker berühmt.
Behandlung des Marfan-Syndroms
Behandlungen für das Marfan-Syndrom sind leider noch nicht entwickelt worden. Die therapeutischen Maßnahmen hängen von den individuellen Ausprägungen der Krankheit ab.
Wenn die SCS-Störungen nicht stark ausgeprägt sind, wird eine konservative Behandlung empfohlen. Bei deutlicher Erweiterung der Aorta ascendens und Aortendissektion, Mitralklappenprolaps, Herzfehlern werden chirurgische Methoden eingesetzt.
Die Sehkraft wird mit Brillen und Kontaktlinsen korrigiert. Falls erforderlich, wird eine chirurgische Behandlung des Glaukoms, des Grauen Stars und der Ersatz der verschobenen Linse durch eine künstliche Linse durchgeführt.
Zur Unterstützung des Bewegungsapparats werden eine Kollagen-Normalisierungstherapie sowie ein Vitamin- und Mineralstoffkomplex verordnet. Schwere Skelettanomalien werden chirurgisch korrigiert.
Klinische Leitlinien schließen körperliche Belastungen, verletzungsanfällige Spiele und hohe Aktivitätsniveaus für Patienten mit der Mutation aus. Zur Unterstützung der Wirbelsäule kann ein Korsett verwendet werden, und zur Stärkung der Muskeln werden Physiotherapie und Massage empfohlen.
Das Leben ist für Menschen mit dieser Genmutation nicht einfach. Doch durch einen verantwortungsvollen Umgang mit ihrer Gesundheit können die Patienten die Symptome der Krankheit verringern und schwerwiegende Komplikationen verhindern.
Was sollten Ärzte über das Marfan-Syndrom wissen?
Da es sich um eine komplexe genetische Erkrankung handelt, ist es für Ärzte wichtig, die wichtigsten Ursachen, Symptome, diagnostischen Grundsätze und gefährlichen Komplikationen zu kennen und zu verstehen. Diese lassen sich wie folgt kurz zusammenfassen:
- Die Ursache ist eine autosomal dominante Genmutation mit gestörter Synthese von Fibrillin-1 im Körper;
- die Symptome sind unterschiedlich, die Klinik ist sehr unterschiedlich, aber es sollte immer auf die Gliedmaßen des Patienten, den Zustand der Linse und der Aorta geachtet werden;
- Auch die Komplikationen sind unterschiedlich, aber die schwerwiegendste ist die Aortendissektion;
- Das Marfan-Syndrom kann nur durch einen Gentest genau diagnostiziert werden.
Strukturelle Anomalien des Skeletts, der Augen und des Herz-Kreislauf-Systems werden durch bildgebende Diagnostik festgestellt, und es sollten sofort Medikamente aus der Gruppe der Betablocker verschrieben werden, um eine Aortenruptur zu verhindern.
Symptome aus verschiedenen Organen und Systemen
Auf der kardiovaskulären Seite werden bei den Patienten am häufigsten ein Klappenprolaps und ein Aortenaneurysma diagnostiziert. Wenn die Aortenwurzel und der aufsteigende Teil der Aorta pathologisch verändert sind, kann dies zu schwerwiegenden Komplikationen führen. Eine Schädigung der Aortenintima tritt meist dort auf, wo die Belastung der Aorta besonders hoch ist. Es kommt zu einer allmählichen Erweiterung des Gefäßes oder zu einer plötzlichen Ruptur des Koronarsinus.
Eine Aortenwurzeldilatation wird bei der Hälfte der pädiatrischen Patienten und bei etwa 60-80 % der erwachsenen Patienten diagnostiziert. Eine Aorteninsuffizienz wird durch Auskultation festgestellt, wobei ein diastolisches Geräusch über der Klappe zu hören ist. Wenn die Klappenblätter und -fäden gedehnt sind, kann es zu einem Mitralklappenprolaps kommen, der von einem systolischen Klicken und Geräuschen unterschiedlicher Intensität begleitet wird. Eine bakterielle Endokarditis kann sowohl an der Aorten- als auch an der Mitralklappe auftreten.
Auf der muskuloskelettalen Seite ist ein wichtiges Symptom, dass der Patient groß ist und seine Arme weit über die Höhe abgespreizt sind. Bei näherer Betrachtung ist eine Arachnodaktylie des Daumens zu erkennen, wobei das distale Fingerglied über den Rand der geballten Faust hinausragt. Der Brustkorb ist entweder gekielt und nach außen verlagert oder trichterförmig und nach innen verlagert. Die Gelenke sind oft hypermobil, mit leichten Kontrakturen in den Ellenbogen. Die Knie können nach hinten gebeugt sein, Plattfüße und Wirbelsäulenverkrümmungen wie Kyphose und Skoliose sind häufig. Auch das Unterhautfettgewebe ist schlecht entwickelt.
Die Ektopie der Linse (Subluxation oder Verschiebung nach oben) ist das wichtigste visuelle Zeichen des Marfan-Syndroms. Iridodeneszenz mit Flattern der Iris ist ebenfalls häufig. Wenn die Linse betroffen ist, ist ihr Rand verschoben, was man sehen kann, wenn die Pupillen des Patienten nicht geweitet sind. Eine Myopie mit spontaner Netzhautablösung ist ebenfalls nicht selten.
Im Bereich der Atemwege treten häufig pathologische Manifestationen in Form einer zystischen Lungenerkrankung und eines Spontanpneumothorax auf, der in der Regel ständig wiederkehrt. Infolgedessen leiden die Patienten häufig unter Atemnot und Brustschmerzen.
Hauptanzeichen der Pathologie

Häufig treten die äußeren Anzeichen des Marfan-Syndroms bereits in den ersten Tagen nach der Geburt auf und verstärken sich erst später. Unter den äußeren Anzeichen, die auf eine Pathologie schließen lassen, sind vor allem folgende zu nennen
- Vermehrte Länge der Gliedmaßen und Finger (Dolichostenomelie und Arachnodaktylie);
- Untergewicht mit erhöhter körperlicher Entwicklung des Kindes;
- Länglicher Schädel und längliches Gesicht;
- Schwaches, unterentwickeltes Muskelgewebe, Fehlen von Fettgewebe;
- Ungewöhnlich hohe Gelenkbeweglichkeit;
- Ungeschicklichkeit und Unbeholfenheit in der Bewegung.
Das Marfan-Syndrom führt bei Kindern über vier Jahren zu Veränderungen der Form des Brustkorbs, einer Verkrümmung der Wirbelsäule und der Entwicklung von Plattfüßen.
Zu den häufigsten Augenmanifestationen gehören Myopie, ektopische Linse, Hornhautveränderungen, Strabismus, Iris- und Netzhauthypoplasie. Diese Veränderungen treten oft schon in den ersten Lebensjahren auf, sind bilateral und schreiten im Laufe der Zeit systematisch voran.
Am schwerwiegendsten sind die pathologischen Veränderungen des Herz-Kreislauf-Systems, die unbehandelt bereits in jungen Jahren zum Tod führen können. Dazu gehören Veränderungen der Gefäßwand und verschiedene Fehlbildungen des Herzens und der Herzkranzgefäße. Bei der ungünstigsten Form der Krankheit entwickelt ein Kind bereits im ersten Lebensjahr eine fortschreitende Herzinsuffizienz.
Darüber hinaus können sich die Symptome des Marfan-Syndroms auch in anderen Systemen und Organen zeigen. Die Krankheit kann das Nervengewebe, die Bronchien und die Lunge, die Haut, die Harnwege und den Genitaltrakt betreffen.
Nur ein Arzt kann die Krankheit genau diagnostizieren. Zögern Sie Ihre Konsultation nicht hinaus – rufen Sie an unter +7 (495) 775-73-60
Wie kann man eine Krankheit bei einem Kind genau diagnostizieren?
Derzeit basiert die Diagnose des Marfan-Syndroms auf dem klinischen Bild gemäß den 1995 entwickelten und 2010 verfeinerten Genter Kriterien. Diese beschreiben eine Reihe von Anzeichen einer Pathologie des Skelettsystems, der Sehorgane, des Herzens und der Blutgefäße sowie anderer Systeme und Organe. Um den Grad der Compliance zu bestimmen, erhebt der Arzt die Krankengeschichte (einschließlich der Familiengeschichte), führt eine gründliche Untersuchung des Patienten mit phänotypischen Tests durch und ordnet Labor- und instrumentelle Tests an, die Folgendes umfassen:

- Urinanalyse zur Bestimmung der Glykosaminoglykane;
- Nachweis des DNA-Genotyps;
- EKG und EchoCG zur Feststellung von Herz- und Gefäßanomalien;
- Ultraschalluntersuchung des Herzens;
- Röntgenaufnahmen des Brustkorbs zur Feststellung von Fehlbildungen des Skelettsystems, des Herzens und der Lunge;
- Computertomographie und Magnetresonanztomographie (MRI).
Je nach Bedarf können weitere Tests und Untersuchungen angeordnet werden.
Diagnose des Marfan-Syndroms
Die Diagnose des Marfan-Syndroms kann schwierig sein, da viele Patienten nur bestimmte charakteristische Anzeichen und Symptome und keine spezifischen histologischen oder biochemischen Veränderungen aufweisen. Aufgrund dieser Variabilität beruhen die Diagnosekriterien auf einer Kombination aus klinischen Befunden, Familienanamnese und genetischer Vorgeschichte. (Weitere Informationen zur Diagnose finden Sie in der überarbeiteten Genter Nosologie). Allerdings ist die Diagnose in vielen Einzelfällen des Marfan-Syndroms unklar.
Homocystinurie Klassische Homocystinurie Eine Reihe von Defekten im Methionin-Stoffwechsel führt zu einer Anhäufung von Homocystein (und Homocystein-Dimeren) mit nachteiligen Auswirkungen wie einer Neigung zu Thrombose, Linsenverschiebung und. Lesen Sie mehr Es kann teilweise das Marfan-Syndrom nachahmen, lässt sich aber durch den Nachweis von Homocystein im Urin unterscheiden. Genetische Tests auf Mutationen FBN1 kann bei Menschen, die nicht alle klinischen Kriterien erfüllen, zur Diagnose beitragen, aber es gibt auch Fälle, in denen die Mutation im FBN1-Gen negativ ist FBN1-MUTATION. Pränatale Diagnose durch Mutationsanalyse FBN1 Gen ist aufgrund der schlechten Genotyp/Phänotyp-Korrelation pränatal schwer zu diagnostizieren (es wurden >1.700 verschiedene Mutationen beschrieben).
Standardmäßige bildgebende Untersuchungen des Skelett-, Herz-Kreislauf- und Sehsystems werden durchgeführt, um klinisch relevante strukturelle Anomalien festzustellen, die für die Diagnose von unbedingtem Wert sind (z. B. Echokardiographie zur Feststellung einer Aortenwurzeldilatation).
Zusätzlich zu den Kriterien für die Organsysteme werden eine Familienanamnese (Verwandte im ersten Knie mit Marfan-Syndrom) und eine genetische Anamnese (Vorhandensein von FBN1 Mutation, die das Marfan-Syndrom verursacht) als Hauptkriterien angesehen.
Prognose beim Marfan-Syndrom
Die Fortschritte in der Therapie und die regelmäßige Überwachung haben die Lebensqualität verbessert und die Sterblichkeit verringert. Die Lebenserwartung hat sich von durchschnittlich 48 Jahren im Jahr 1972 auf eine nahezu normale Lebenserwartung derjenigen erhöht, die eine angemessene medizinische Versorgung erhalten. Die Lebenserwartung des Durchschnittspatienten ist jedoch immer noch niedriger, was vor allem auf kardiale und vaskuläre Komplikationen zurückzuführen ist. Diese verkürzte Lebenserwartung kann eine emotionale Belastung für Jugendliche und Familien darstellen.
Die Behandlung des Marfan-Syndroms zielt darauf ab, Komplikationen zu verhindern und zu behandeln.
Bei sehr großen Mädchen kann die Induktion einer vorzeitigen Pubertät im Alter von 10 Jahren mit Östrogenen und Progesteron das Wachstum im Erwachsenenalter einschränken.
Alle Patienten sollten regelmäßig Betablocker (z. B. Atenolol, Propranolol) erhalten, um kardiovaskuläre Komplikationen zu verhindern. Diese Medikamente verringern die Kontraktilität des Herzmuskels und den Pulsdruck und reduzieren das Fortschreiten des Aortenwurzelaneurysmas und das Risiko einer Aortendissektion. Auch Angiotensin-II-Rezeptorblocker können eingesetzt werden.
Bei einem Aortendurchmesser von > 5 cm (bei Kindern kleiner) wird eine prophylaktische Operation empfohlen. Schwangere Frauen haben ein besonderes Risiko für Aortenkomplikationen; eine empfohlene Aortenrekonstruktion sollte vor der Empfängnis besprochen werden. Schwere Klappeninsuffizienzen werden ebenfalls chirurgisch rekonstruiert. Prophylaxe der bakteriellen Endokarditis Die infektiöse Endokarditis (IE) ist eine Infektion des Endokards, die in der Regel bakteriell (häufiger durch Streptokokken oder Staphylokokken) oder durch Pilze verursacht wird. Sie kann sich durch Fieber, Herzgeräusche und Petechien äußern. Lesen Sie mehr Nicht indiziert vor invasiven Eingriffen, außer bei Patienten, denen künstliche Herzklappen implantiert wurden oder die an einer infektiösen Endokarditis erkrankt sind (Verfahren, die eine prophylaktische Antibiotikatherapie bei Patienten mit hohem Endokarditis-Risiko erfordern, in den Vereinigten Staaten verwendet Verfahren, die eine prophylaktische Antibiotikatherapie bei Patienten mit hohem Endokarditis-Risiko erfordern, in den Vereinigten Staaten verwendet. i Empfohlene Prophylaxe gegen Endokarditis bei zahnärztlichen Eingriffen und Manipulationen der oberen Atemwege* Empfohlene Prophylaxe gegen Endokarditis bei zahnärztlichen Eingriffen und Manipulationen der oberen Atemwege ).
Was sind die Symptome des Marfan-Syndroms?

Diese Krankheit ist dadurch gekennzeichnet, dass mehrere Systeme gleichzeitig betroffen sind. Sie kann mit einer Vielzahl von klinischen Symptomen einhergehen. Der Zeitpunkt des Auftretens und der Schweregrad der Symptome können von Patient zu Patient sehr unterschiedlich sein.
- Diese Menschen haben vor allem bestimmte äußere Merkmale. Sie zeichnen sich durch ihre hohe Statur aus. Ihr Rumpf ist relativ kurz, während ihre Gliedmaßen überproportional lang sind. Das Unterhautfettgewebe ist unterentwickelt und der Muskeltonus ist gering. Das Gesichtsskelett ist langgestreckt und eingeengt.
- Es kann auch mit verschiedenen Skelettdeformationen einhergehen, wie z. B. einer kielförmigen Vorwölbung des Brustkorbs, einer Krümmung der Wirbelsäule und so weiter.
- Das auffälligste Merkmal des Marfan-Syndroms ist die Schädigung des Herz-Kreislauf-Systems. Abnorme Aortenläsionen wie Aortenaneurysmen oder Herzklappen werden bei diesen Patienten häufig festgestellt. Verschiedene angeborene Herzfehler sind möglich. Ein häufiges Symptom ist ein abnormaler Herzrhythmus.
- In schweren Fällen kann die Herzfehlfunktion rasch fortschreiten und in kurzer Zeit zu einem Kreislaufversagen führen.
- Darüber hinaus werden häufig Augenstörungen beobachtet, die sich in Form von Kurzsichtigkeit, Linsenektopie, Schielen und anderem äußern können.
Auch andere innere Organe, wie z. B. das Atmungssystem, können betroffen sein. Ein häufiges Anzeichen für eine Beteiligung der Atemwege ist der Spontanpneumothorax, ein Merkmal des Marfan-Syndroms, wie Forscher der Staatlichen Pädiatrischen Medizinischen Universität St. Petersburg in einer 2022 veröffentlichten Arbeit nachgewiesen haben.
Wie erfolgt die Behandlung des Marfan-Syndroms?
Für die Behandlung des Marfan-Syndroms in seiner Gesamtheit wurde keine spezifische Therapie entwickelt. Alle Behandlungen sind symptomatischer Natur.
In erster Linie werden Maßnahmen ergriffen, um die normale Herz-Kreislauf-Funktion wiederherzustellen und zu erhalten. Bei solchen Patienten können Betablocker, Angiotensin-Converting-Enzyme-Hemmer usw. verabreicht werden. Bei Bedarf werden chirurgische Eingriffe wie der Ersatz der Mitralklappe durch eine Prothese vorgenommen.
Das Sehvermögen kann durch die Anpassung einer Brille oder durch chirurgische Eingriffe wie die Laserentfernung des Grauen Stars korrigiert werden.
Darüber hinaus werden Präparate zur Steigerung der Kollagenproduktion, Stoffwechselprozesse und Vitaminkomplexe eingesetzt, um die Kollagenproduktion zu erhöhen.
Wie äußert sich das Marfan-Syndrom?
Zunächst einmal fällt das besondere Aussehen des Patienten auf. Die Betroffenen sind groß, haben lange Arme und Beine, die in keinem Verhältnis zu anderen Körperteilen stehen, und dünne, längliche Finger. Das Unterhautfettgewebe ist unterentwickelt und der Muskeltonus ist reduziert. Das Gesichtsskelett ist lang und schmal. Charakteristisch sind auch ein hochgewölbter Gaumen und verschiedene Okklusionsanomalien.
Häufig zeigen Patienten mit Marfan-Syndrom eine übermäßige Beweglichkeit der Gelenke und verschiedene strukturelle Veränderungen der Wirbelsäule und des Brustkorbs.
Es liegen immer Anomalien in der Struktur des Herzens und der Blutgefäße vor. Aortendilatation, Aortenaneurysmen und verschiedene angeborene Herzfehler, wie z. B. ein Ventrikelseptumdefekt, können bei dieser Krankheit auftreten. Die Krankheit wird häufig von Herzrhythmusstörungen begleitet.
Typischerweise äußert sich das Marfan-Syndrom durch verschiedene Augenanomalien. Im Jahr 2012 führten Forscher des Tashkent Paediatric Medical Institute eine Studie durch, in der sie feststellten, dass Augenschäden bei 50-80 % der Patienten auftreten, oft als eines der ersten Anzeichen der Krankheit.
Zu den klinischen und morphologischen Augenanomalien beim Marfan-Syndrom gehören Myopie, ektopische Linse, Unterentwicklung der Iris, Strabismus usw.
Darüber hinaus können auch andere innere Organe wie das zentrale Nervensystem, das bronchopulmonale System, die Haut usw. betroffen sein.
Behandlungsgrundsätze für das Marfan-Syndrom

Aufgrund des angeborenen Charakters des Marfan-Syndroms ist eine vollständige Heilung nicht möglich. Die gesamte Therapie ist symptomatisch und zielt eher darauf ab, das Fortschreiten der Krankheit zu stoppen.
Zur Verbesserung des Herz-Kreislauf-Systems können Betablocker, Angiotensin-Converting-Enzyme-Hemmer usw. verschrieben werden. Bei strukturellen Veränderungen des Herzens oder der Gefäße kann eine chirurgische Behandlung, z. B. eine rekonstruktive Aortenoperation oder ein Klappenersatz, durchgeführt werden.
Augenkrankheiten können mit Brillen oder Kontaktlinsen, Operationen zur Beseitigung von grünem oder grauem Star usw. korrigiert werden.
Schwerwiegende Veränderungen des Bewegungsapparats können ebenfalls chirurgische Eingriffe wie Brustwandrekonstruktionen, Operationen zur Stabilisierung der Wirbelsäule usw. erforderlich machen.
Lesen Sie mehr:- Marfan-Syndrom bei Neugeborenen.
- Marfan-Syndrom.
- Marfan-Syndrom – Zusammenfassung.
- Ehlers-Danlo-Syndrom.
- Behandlung des Kurzbeinsyndroms.
- Wie man einen Klumpfuß bei einem Kind korrigiert.
- Anatomie der Beckenknochen.
- Ektrodaktylie.