Charcot-Marie-Tooth-Neuromyotrophie (CMT) bezeichnet eine Gruppe progressiver, chronischer, vererbter Polyneuropathien, zu denen das Roussi-Levy-Syndrom, die hypertrophe Degenerativ-Sott-Neuropathie, die Refsum-Krankheit und andere, seltenere Erkrankungen gehören.

- Neurale Atheropathie: Ursachen, Symptome, Diagnose
- Internationale Klassifikation der neuralen Atheropathien
- Hintergrundinformationen.
- Ursachen des Roussi-Levi-Syndroms
- Ursachen
- Pathogenese
- Symptome der Refsum-Krankheit
- Diagnose
- Pathogenetische Aspekte der Charcot-Marie-Tooth-Krankheit und ihre Symptome
- Klassifizierung der Charcot-Marie-Tooth-Krankheit
- Wirkung
- Häufig gestellte Fragen
- Klassifizierung
- Diagnose
- Klinisches Bild
- Diagnose
- Behandlungsansatz
- Vorbeugung
Neurale Atheropathie: Ursachen, Symptome, Diagnose
Syndrome der Demyelinisierung von Nervenfasern mit autosomal-dominanter Vererbung durch das X-Chromosom treten bei 2 Personen pro hunderttausend Einwohner auf. Die Krankheit ist nicht typisch für das Gebiet der Russischen Föderation, da es auf dem heimischen Territorium kein Reservoir für die Existenz von Erregern gibt.
Der chronische Verlauf der Krankheit geht mit einem allmählichen Fortschreiten der Muskellähmung und Lähmung einher. Die Pathologie geht mit Veränderungen der Muskulatur einher, die zunächst in den Beinen und in zweiter Linie in den Armen allmählich atrophieren. Die Faszikulationen werden durch eine Unterbrechung der Cholinesteraseübertragung der Nervenfasern infolge einer infektiösen Läsion verursacht. Die unterstützende Behandlung zielt auf die Wiederherstellung der antioxidativen Funktion und der metabolischen Dysfunktion ab.
Die reduzierte Nährstoffzufuhr ist durch atrophische Veränderungen der Muskulatur und den Verlust der kontraktilen Funktion gekennzeichnet. Die pathogenetischen Mechanismen sind auf die Zerstörung der motorischen Neuronen des Rückenmarks, der Rumpfstrukturen und der Vorderhörner des Mittelhirns zurückzuführen.

Multifokale Leukoenzephalopathie im MRT
Internationale Klassifikation der neuralen Atheropathien
Eine Reihe von Pathologien, die mit motorischen und sensorischen Beeinträchtigungen einhergehen, werden als hereditäre Amyotrophien klassifiziert. Die internationale Klassifikation ICD 10 kodiert diese Nosologie als ‚G12‘. Die Vererbung verhindert eine ätiologische Behandlung. Ein Syndrom gemeinsamer Störungen mit Taubheit, Retinitis pigmentosa und Ataxie wird mit unterstützenden Medikamenten behandelt.
- Charcot-Marie-Tooth;
- Dejerin-Sotta – hypertrophe Läsionen der Nervenfaserhüllen verursachen eine allmähliche Zunahme der klinischen Symptome aufgrund einer sensomotorischen Neuropathie;
- Degenerative Veränderungen des Rumpfes, der Hörner des Rückenmarks treten in drei klinischen Formen auf – spät, früh und angeboren. Die Nosologie wird als ’spinale Muskelatrophie nach Werdnig-Hoffmann‘ bezeichnet;
- Die Zerstörung der Motoneuronen mit anschließender spinaler Hornatrophie ist durch Muskelzuckungen gekennzeichnet. Diese Pathologie wird als ‚Kullberg-Wehlander-Atrophie‘ bezeichnet;
- Andere erbliche Amyotrophien sind die neurogene maligne, die skapulohumerale und die New-England-Amyotrophie.
Nach der Internationalen Klassifikation der Krankheiten sollten Symptome mit ähnlichen klinischen Merkmalen unterschieden werden. Verwandte Erscheinungsformen werden nach unterschiedlichen Mustern behandelt, so dass eine Differenzialdiagnose erforderlich ist.
- Der erste Typ geht mit veränderten Eigenschaften der Nervensignalübertragung inmitten der Zerstörung der Myelinscheiden einher;
- Bei der zweiten Variante bleibt die Impulsivität bestehen, aber es wird eine axonale Schädigung der Schwann-Zellen festgestellt.
Laut Wikipedia ist eine frühzeitige Überprüfung der morphologischen Formen der Krankheit notwendig, bevor sich Komplikationen entwickeln.
Hintergrundinformationen.
Das Roussi-Levy-Syndrom ist nach dem französischen Neurologen Levy und dem Pathologen Roussay benannt, die es 1926 beschrieben. Die Autoren beschrieben das Syndrom als familiäre areflektische Dystasie mit langsamer Progression. Ataxie und Sehnenareflexie sind die primären klinischen Manifestationen des Syndroms, woraus sich auch sein anderer Name ableitet: Ataxie-Areflexie-Syndrom. Nach der 10. Revision der internationalen Klassifikation wird das Russi-Levi-Syndrom in die Gruppe der hereditären motorisch-sensorischen Neuropathien (HMSN) eingeordnet. Allerdings besteht unter Klinikern kein eindeutiger Konsens darüber, wie diese Pathologie zu klassifizieren ist. Entsprechend der Symptomatik stufen einige Neurologen das Roussi-Levy-Syndrom als eine klinische Form ein, die zwischen der Charcot-Marie-Tooth-Krankheit und der Friedreich-Ataxie liegt; andere betrachten es als eine eigene klinische Variante der spinalen Amyotrophie. Trotz der genetischen Beweise, die für die letztgenannte Aussage sprechen, behandeln viele Neurologen das Roussi-Levy-Syndrom immer noch als eigenständige Nosologie.
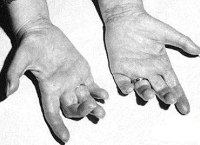
Ursachen des Roussi-Levi-Syndroms
Das familiäre Auftreten der Krankheit lässt auf einen erblichen Ursprung schließen. Die Roussi-Levy-Neuropathie wurde früher in Familien mit wiederkehrenden Fällen der Charcot-Marie-Tooth-Krankheit gefunden. Die in den letzten Jahren durchgeführten molekulargenetischen Untersuchungen haben ergeben, dass die genetische Grundlage des Syndroms eine Anomalie auf Chromosom 17 (Locus 17p11.2) in Form von Verdoppelungen einzelner Gene und Punktmutationen im Myelinprotein-Null-Gen (MPZ) ist. Aufgrund dieser Feststellung kann das Roussi-Levi-Syndrom als phänotypische Form der neuronalen Charcot-Marie-Tooth-Amyotrophie (NMSN-Variante IA) angesehen werden.
Dieses Syndrom wird autosomal dominant vererbt. Morphologisch zeigen sich degenerative Veränderungen der hinteren Spinalwurzeln und der peripheren Nervenstämme in Form einer mäßigen Demyelinisierung und Reduktion der Nervenfasern; dystrophische Veränderungen der Pyramiden- und Kleinhirnbahnen, die in den seitlichen Säulen der Wirbelsäule verlaufen; Degeneration der hinteren Säulen und Atrophie der Muskelfasern der Skelettmuskulatur.
Ursachen
Bis heute verfügt die praktische Neurologie als Wissenschaft nicht über zuverlässige Daten zur Ätiologie und Pathogenese der Nervenamyotrophie. Studien haben gezeigt, dass 70-80% der CMT-Patienten, die sich einem Gentest unterzogen haben, eine Duplikation einer bestimmten Region des Chromosoms 17 aufweisen. Die neuronale Charcot-Marie-Tooth-Amyotrophie kann verschiedene Formen annehmen, die möglicherweise auf Mutationen in verschiedenen Genen zurückzuführen sind. So fanden Forscher heraus, dass die durch eine Mutation im Gen für das mitochondriale Protein MFN2 verursachte Form der CMT zu einer Verklumpung der Mitochondrien führt, die ihre Bewegung entlang des Axons stört.
Die Charcot-Marie-Tooth-Krankheit ist durch einen autosomal dominanten Erbgang mit einer Penetrationsrate von 83 % gekennzeichnet. Es gibt auch Fälle von autosomal-rezessivem Erbgang.
Pathogenese
Bei den meisten Formen der CMT wurde eine Schädigung der Myelinscheide der peripheren Nervenfasern festgestellt; seltener sind die Axone, die axialen Zylinder, die durch das Zentrum der Nervenfaser verlaufen, betroffen. Degenerative Veränderungen betreffen auch die vorderen und hinteren Rückenmarkswurzeln, die Vorderhornneuronen, die Gaul’schen Bahnen (spinalen tiefsensiblen Leitungsbahnen) und die Clark’schen Säulen, die zu den hinteren Bahnen des Rückenmarks gehören.
In zweiter Linie entwickelt sich infolge der Dysfunktion der peripheren Nerven eine Muskelatrophie, an der bestimmte Gruppen von Myofibrillen beteiligt sind. Das weitere Fortschreiten der Krankheit ist durch eine Verschiebung der Sarkolemmkerne, eine Hyalinisierung der veränderten Myofibrillen und eine Hypertrophie des interstitiellen Bindegewebes gekennzeichnet. In der Folge führt die zunehmende hyaline Degeneration der Myofibrillen zu deren Zusammenbruch.
Symptome der Refsum-Krankheit
Multiple Läsionen der peripheren Nervenstämme führen zu den Symptomen der Polyneuropathie: Taubheitsgefühl in Händen und Füßen, Parästhesien, verminderte Schmerzwahrnehmung und tiefe (propriozeptive, vibratorische) Sensibilität. Die Polyneuropathie ist sensomotorisch bedingt. Die motorischen Störungen beginnen hauptsächlich mit einer Parese der Streckmuskeln der Füße, dann breitet sich die schlaffe Parese auf die distalen Teile der Beine und Arme aus, begleitet von einer Hypotrophie der Muskeln der Füße, Unterschenkel, Hände und Unterarme. Charakteristisch ist eine Kleinhirnataxie mit unsicherem Gang, Hypermetrie und veränderter Handschrift. Beim Sehen werden eine verminderte Sehschärfe, Hämeralopie, verengte Gesichtsfelder und in einigen Fällen Katarakte beobachtet. Manchmal liegt eine Photophobie vor.
Häufig geht die Refsum-Krankheit mit Hörverlust, Geruchsverlust (Anosmie), hängenden Oberlidern und okulomotorischen Störungen einher. Die Haut ist trocken und schuppig. Der Schweregrad der Ichthyose variiert von leichter Hyperkeratose an Füßen und Händen mit kleinen weißen Krusten bis hin zu schwerer lamellärer Ichthyose. Einige Patienten haben begleitende Skelettdysplasien: Krümmung der Wirbelsäule (Skoliose), Fußdeformitäten (Hohlfuß, Krümmung und Verkürzung der Zehen).
Diagnose
Die Diagnose kann anhand einer Blut- und einer Urinuntersuchung gestellt werden, bei denen die Phytansäurekonzentration bestimmt wird. Dieser Wert steigt bei der Refsum-Krankheit auf 800 µmol/l an, während der Normalbereich unter 19 µmol/l liegt. Bei allen Patienten wird ein Augenarzt hinzugezogen. Die Untersuchung der Sehschärfe zeigt eine verminderte Sehschärfe, die Perimetrie eine konzentrische Verengung der Gesichtsfelder und die Ophthalmoskopie Anzeichen einer Sehnervenatrophie und polymorphe degenerative Veränderungen der Netzhaut.
Die Elektroneuromyographie kann den polyneuropathischen Charakter des peripheren Nervensystems feststellen und Anomalien der Vorderhornmotoneuronen aufdecken. Erhöhte Albuminwerte werden im Liquor diagnostiziert. Störungen des Geruchsinns werden durch Olfaktometrie festgestellt. Hörstörungen werden durch Beratung mit einem Audiologen und durch Audiometrie festgestellt.
Das Vorhandensein von Knochendeformationen ist die Grundlage für die Verschreibung einer Röntgenaufnahme der Wirbelsäule, eines CTs oder einer Röntgenaufnahme der Füße, gefolgt von einer Konsultation eines Orthopäden. Zur Beurteilung des Herzens wird ein EKG durchgeführt und ein Kardiologe hinzugezogen. Eine genealogische Studie wird durchgeführt, um die Art der Vererbung der Pathologie zu bestimmen. Die endgültige Diagnose kann durch eine DNA-Analyse bestätigt werden.
Bei der Diagnose wird die Refsum-Krankheit von der Charcot-Marie-Tooth-Amyotrophie, Polyneuropathien anderen Ursprungs (Degenerina-Sotta-Syndrom, diabetische Neuropathie, Russ-Levy-Syndrom, paraneoplastische Neuropathie usw.) und der Friedreich-Ataxie unterschieden.
Pathogenetische Aspekte der Charcot-Marie-Tooth-Krankheit und ihre Symptome
Die Pathogenese der Charcot-Marie-Tooth-Krankheit beruht auf einer Nervenschädigung, die zu einer sekundären Muskelatrophie führt. Die schnell verlaufenden motorischen Nerven, die die am stärksten beanspruchten Muskeln der Gliedmaßen – die Fuß- und Unterschenkelmuskeln – versorgen, sind am häufigsten geschädigt. Die Muskeln der Hände und Unterarme sind etwas weniger und später betroffen. Auch die sensorischen Nerven sind betroffen, was zu einem Verlust der Empfindlichkeit für Berührung, Schmerz und Temperatur in den Gliedmaßen führt.
Die Charcot-Marie-Tooth-Krankheit manifestiert sich in der Regel zwischen dem 10. und 20. Zunächst kommt es zu einer symmetrischen Schwäche der unteren Gliedmaßen, die sich in einem charakteristischen Schritt oder einem so genannten ‚Schlingergang‘ äußert. Dann beginnen sich die Füße zu verdrehen und zu verformen, das Fußgewölbe verbreitert sich und es entsteht ein Hohlfuß. Mit fortschreitendem Muskelschwund nehmen die Füße das Aussehen von umgedrehten Flaschen an.
Nach und nach sind auch die Hände betroffen: Die Atrophie der Hand lässt sie wie eine Affenpfote aussehen. Darüber hinaus ist die Sensibilität, insbesondere die oberflächliche Empfindung, beeinträchtigt. Manchmal kommt es zu einer Zyanose der betroffenen Gliedmaßen und einer Schwellung der Extremitäten.
Die Charcot-Marie-Tooth-Krankheit hat einen langsamen Verlauf. Manchmal vergehen 10 Jahre zwischen Manifestation und Atrophie der Hände. Auch wenn sich eine Atrophie entwickelt, können die Patienten noch lange arbeiten und sich selbst versorgen. Bei angemessener Pflege bleibt die Lebenserwartung auf dem Niveau der Allgemeinbevölkerung.
Klassifizierung der Charcot-Marie-Tooth-Krankheit
Die genetische Klassifizierung ist sehr breit gefächert, da etwa 40 Mutationen, die mehr als 20 verschiedene Gene betreffen, der Entstehung der Krankheit zugrunde liegen. Es gibt drei Haupttypen der Charcot-Marie-Tooth-Krankheit, die auf morphologischen Merkmalen und elektromyografischen Daten beruhen:
- Demyelinisierender Typ. Kennzeichnend ist die Zerstörung der Myelinscheide und die daraus resultierende Verringerung der Impulsgeschwindigkeit (IPV) entlang des Medianusnervs;
- Axonaler Typ. Der Nervus medianus ist durch einen normalen oder leicht reduzierten SPI gekennzeichnet, da hauptsächlich Axone geschädigt sind;
- intermediärer Typ. Die Impulsübertragungsgeschwindigkeit ist grenzwertig.
Diese Klassifizierung ist nützlich, um die Suche nach einer genetischen Diagnose einzugrenzen, da einige Mutationen unterschiedliche klinische Manifestationen haben.
Wirkung
Schwere Deformitäten können nur chirurgisch korrigiert werden, da sie eine unabhängige Bewegung praktisch unmöglich machen. Ein chirurgischer Eingriff ist jedoch nur dann angezeigt, wenn konservative Methoden unwirksam sind.
Die häufigsten Eingriffe sind Osteotomien – Osteotomien des Fersenbeins oder Osteotomien der Mittelfußknochen. In extremen Fällen wird eine Arthrodese durchgeführt, bei der das Gelenk nach der Operation vollständig ruhiggestellt bleibt.
Vereinbaren Sie online einen Beratungstermin, um eine Operation bei Morbus Charcot so lange wie möglich zu vermeiden oder hinauszuzögern. Unsere Ärzte informieren Sie umfassend über alle auftretenden Probleme und Symptome. Unsere Neurologen sind 24 Stunden am Tag für Sie da.
Häufig gestellte Fragen
Das Charcot-Marie-Tooth-Syndrom wird häufiger bei Männern als bei Frauen diagnostiziert.
Der wichtigste Faktor ist das Auftreten der Charcot-Marie-Tooth-Muskelatrophie bei engen Familienmitgliedern. Wenn beide Eltern Träger des autosomal rezessiven Gens sind, besteht eine 25-prozentige Chance, ein Kind mit dieser Pathologie zu bekommen. Es kann aber auch sein, dass das Kind nur Träger des Gens ist, d. h. dass es im Laufe seines Lebens keine Symptome zeigt: Dieses Risiko ist mit 50 % höher. Bei der X-chromosomalen Vererbung wird das Risiko, dass das Gen von der Mutter auf den Sohn übertragen wird, auf 50 % geschätzt, aber das Kind wäre nur Träger. Bei der Geburt eines Mädchens kann die Krankheit nicht weitergegeben werden, aber ihre Söhne, d. h. die Enkelkinder der Patientin Null, können das defekte Gen erben. Und sie werden bereits das Charcot-Marie-Tutte-Syndrom entwickelt haben.
Dies geschieht vor allem als Folge von Infektionen wie Masern, Röteln, infektiöser Mononukleose, Pharyngitis oder auch einer Erkältung. Erkältungen, Kopf- und Wirbelsäulenverletzungen und Vitaminmangel können ebenfalls begünstigende Faktoren sein.
Klinische Manifestationen treten in der Regel bei Kindern über 10 Jahren auf, seltener zwischen 16 und 30 Jahren.
Klassifizierung
Klinische und genetische Klassifizierung der hereditären motorischen Neuropathien
Autosomal-dominante hereditäre motorisch-sensorische Neuropathie, Typ I:
– Verursacht durch Punktmutationen im Myelin-Gen PMR-22.
Typ I B: Verursacht durch Punktmutationen im Rho-Myelin-Gen.
Typ I C: mit einem nicht identifizierten Gendefekt.
Autosomal rezessive hereditäre motorisch-sensorische Neuropathie, Typ I: mit nicht identifiziertem Gendefekt.
Autosomal-dominante hereditäre motorisch-sensorische Neuropathie, Typ II: mit nicht identifiziertem Gendefekt.
Autosomal rezessive hereditäre motorisch-sensorische Neuropathie, Typ II: mit nicht identifiziertem Gendefekt.
Hereditäre motorisch-sensorische Neuropathie, Typ III:
– verursacht durch Punktmutationen im Myelin-Gen PMR-22;
– verursacht durch Punktmutationen im Myelin-Rho-Gen.
X-chromosomal vererbte sensorisch-motorische Neuropathie:
– verursacht durch Punktmutationen und Deletionen in Connexin 32;
– mit einem nicht identifizierten Gendefekt.
Komplexe Formen hereditärer motorisch-sensorischer Neuropathien:
– Mit Sehnervenatrophie und Taubheit;
Diagnose
Diagnostische Kriterien
Beschwerden und Anamnese: Muskelermüdung, Muskelatrophie, Schmerzen vom ‚polyneurischen Typ‘ in den distalen Gliedmaßen, Skelettdeformitäten, Gangstörungen, Verzögerung der motorischen Entwicklung, progressiver Verlauf.
Anamnese: bei der Charcot-Marie-Krankheit: autosomal rezessiver Erbgang, Beginn im ersten Lebensjahrzehnt.
Bei der Degerin-Sott-Krankheit (hereditäre motorisch-sensorische Neuropathie, Typ III): autosomal rezessiver Erbgang, Beginn in den ersten beiden Lebensjahren, verzögerte motorische Entwicklung, Schwäche und Atrophie der distalen Gliedmaßen mit Progression zu proximalen Läsionen, Ataxie, schneller Verlauf.
Die hereditäre motorisch-sensorische Neuropathie vom Typ I ist durch einen autosomal-dominanten Vererbungstyp gekennzeichnet, Krankheitsbeginn hauptsächlich im 1. bis 2.
Der Typ II der hereditären motorisch-sensorischen Neuropathie ist durch einen autosomal-dominanten Vererbungstyp gekennzeichnet, Krankheitsbeginn im zweiten Lebensjahrzehnt, fehlende oder nur gering ausgeprägte Atrophie der distalen Gliedmaßen, relativ milder Verlauf.
Die hereditäre motorisch-sensorische Neuropathie Typ II ist gekennzeichnet durch einen autosomal-rezessiven Vererbungstyp, Debüt in jungen Jahren, ausgeprägte Atrophie und Schwäche der distalen Gliedmaßen, Deformierungen der Hände und Füße, rasches Fortschreiten des Verlaufs.
Körperliche Untersuchung: Neurologischer Zustand – Schmerzen (schmerzhaft, einschießend, verkrampfend) in den distalen Gliedmaßen vor dem Hintergrund einer Hypoästhesie, mit allmählich einsetzender Schwäche, Atrophie in den distalen Gliedmaßen, Verringerung und Verlust des Achillesreflexes mit fortschreitender Progredienz – karpo-radial; Gangstörung ähnlich dem ‚Gockeln‘, Treten.
Die Atrophie tritt in den distalen Gliedmaßen auf, mit anschließender (fortschreitender) Atrophie in den proximalen Gliedmaßen. Deformierter Klumpfuß oder Friedreich-Fuß in Form eines Hohlfußes mit hohem Fußgewölbe und Streckung der Großzehenglieder. Beginnt die Krankheit im Kindesalter, kommt es zu einer Verzögerung der motorischen Entwicklung.
Klinisches Bild
Der erste Ausbruch der Krankheit beginnt in der Regel im Jugendalter. Zunächst verspürt der Patient eine leichte Schwäche in den unteren Gliedmaßen, die allmählich zunimmt.
Mit der Zeit kommt es zu einer zunehmenden Müdigkeit bei geringer körperlicher Anstrengung und zu Muskelschmerzen. Es wird schwierig, sich zu bewegen, da die Beine hochgehoben werden müssen, was aufgrund der Entwicklung einer asymmetrischen Atrophie nur schwer möglich ist. Die Beine, einschließlich der Füße, können beginnen, taub zu werden. Zehn Jahre nach dem Auftreten der ersten Symptome wird bei den Patienten eine Muskelschwäche diagnostiziert.
Die symmetrische Muskelschwäche führt bei allen Betroffenen zur Diagnose des Pferdefußes. An den oberen Gliedmaßen hingegen sind weniger als die Hälfte der Betroffenen von den Symptomen der Lou-Gehrig-Krankheit betroffen.
Bei der Charcot-Krankheit sind die Hauptsymptome ein verminderter und vollständiger Verlust der Wadenmuskelkontraktion, des Kniescheibenreflexes und dann des Knöchelreflexes. Außerdem treten Empfindungsstörungen in den oberen und unteren Gliedmaßen auf, und der Gang der Betroffenen verändert sich.
‚Ich habe die Marie-Charco-Tooth-Krankheit, wie lange lebe ich mit dieser Krankheit?
Es gibt keine genauen Angaben zur Lebenserwartung bei amyotropher Sklerose. Manche Patienten können ihr Leben wie normale Menschen leben, andere nicht. Bei der primären amyotrophen Lateralsklerose ist nur das obere Motoneuron betroffen, und diese Form der Charcot-Krankheit ermöglicht es den Patienten, lange genug zu leben.
Die kürzeste Lebenserwartung hat die progressive bulbäre Lähmung. Ein Mensch mit dieser Form der Pathologie lebt zwischen sechs Monaten und drei Jahren. Die Lebenserwartung bei progressiver Muskelatrophie liegt im Durchschnitt bei fünf bis 10 Jahren.
Diagnose
Der erste Schritt besteht darin, die Krankheit korrekt von der progressiven Muskelatrophie, der progressiven bulbären Lähmung, der Myopathie-Nevinna und der Polyneuropathie bei Kryoglobulinämie zu unterscheiden.
Die Charcot-Krankheit führt häufig zum Verlust der Sprache. Solange eine Person noch sprechen kann, lohnt es sich daher, ihr das Lippenlesen beizubringen.
Die Technik geht mit der Zeit und fast jede Familie hat einen Computer, einen Laptop oder ein Telefon. Mit einer speziellen Software kann der Patient einen Text in das Gerät eingeben, das ihn dann vorliest.
Manche Familien haben diese Möglichkeit nicht oder der Patient ist sehbehindert. In diesem Fall können speziell angefertigte Karten verwendet werden. Wenn vorher festgelegt wurde, was das Zeichen bedeuten soll, ist es wichtig, es deutlich zu zeichnen, damit der Patient auf die Karte zeigen oder sie vorzeigen kann, um seinen Wunsch zu äußern.
Darüber hinaus kann man versuchen, selbst zu erraten, was der Patient möchte, oder Fragen verwenden, auf die es nur eine Ja- oder Nein-Antwort gibt.
Es sei darauf hingewiesen, dass es bis heute keine wirksame Behandlung für die Charcot-Krankheit gibt. Grundsätzlich zielen alle Therapien darauf ab, das weitere Fortschreiten der Krankheit zu stoppen und die aufgetretenen Symptome so weit wie möglich zu beseitigen.
Bisher gibt es eine kleine Anzahl von Medikamenten, die das Leben eines Charcot-Patienten verlängern – eines davon ist Rilutec. Bei diesem Medikament handelt es sich im Wesentlichen um einen Blocker von Tetrodotoxin (ein starkes, natürlich vorkommendes Nicht-Protein-Gift) über empfindliche Natriumkanäle. Rilutec wird ausschließlich nach Rücksprache mit Ihrem Arzt verschrieben, der sich Ihre Krankengeschichte genau ansehen sollte. Damit soll sichergestellt werden, dass keine anderen Krankheiten vorliegen, bei denen das Medikament absolut kontraindiziert ist.
Die Tagesdosis beträgt 100 mg lebenslang. Das Medikament sollte abgesetzt werden, wenn Sie bei den Tests, die während der Einnahme von Rilutec oder seinen Analoga regelmäßig durchgeführt werden, Anomalien feststellen. Das Medikament wird normalerweise in Tablettenform verkauft.
Behandlungsansatz
Die Behandlung ist auf die Symptome der Charcot Marie Tut Neuroma Amiotrophie zugeschnitten. Die Interventionen sind komplex und lebenslang.
Es ist zu beachten, dass es in der Medizin keine wirksameren Therapien gibt. Es werden nur Techniken angewandt, die dazu beitragen, die Beschwerden des Patienten zu lindern und seine Lebensqualität zu verbessern.
Es ist wichtig, die funktionelle Koordination und Mobilität des Patienten zu optimieren. Therapeutische Maßnahmen sollten darauf abzielen, geschwächte Muskeln vor Verletzungen und verminderter Sensibilität zu schützen.
Die Angehörigen des Patienten müssen ihn im Kampf gegen die Krankheit in jeder Hinsicht unterstützen. Schließlich findet die Behandlung nicht nur in medizinischen Einrichtungen, sondern auch zu Hause statt.
Alle empfohlenen Maßnahmen müssen strikt und täglich durchgeführt werden. Andernfalls werden die Ergebnisse der Behandlung ausbleiben.
Die Behandlung der Amyotrophie umfasst eine Reihe von Techniken:
- physiotherapeutische Verfahren;
- Beschäftigungstherapie;
- eine Reihe von körperlichen Übungen;
- spezielle Fußstützvorrichtungen
- Orthopädische Fußbettungen zur Korrektur eines deformierten Fußes;
- Fußpflege;
- regelmäßige Konsultationen mit dem behandelnden Arzt;
- Einsatz der orthopädischen Chirurgie;
- Injektionen von B-Vitaminen;
- Verabreichung der Vitamine E, A und C.
- Bei amyotrophen Läsionen wird eine spezielle Diät zubereitet.. Eine Diät, die reich an Eiweiß und Kalium ist, sowie eine erhöhte Zufuhr von Vitaminen ist angezeigt.
- Im regressiven Verlauf der Krankheit wird parallel zu den oben genannten Maßnahmen eine Schlammbadbehandlung durchgeführt. Empfohlene Schlamm-, Rhodonium-, Eruptiv-, Sulfid- und Schwefelwasserstoffbäder. Zur Stimulierung der peripheren Nerven wird auch eine Elektrophoresebehandlung durchgeführt.
- Bei Gelenkfehlstellungen und Verformungen des Skelettsystems Eine Korrektur durch einen Orthopäden ist angezeigt..
Vorbeugung
Die Vorbeugung ist. Konsultation eines Genetikers. Impfungen gegen Polio und durch Zecken übertragene Enzephalitis sollten sofort erfolgen.
Das Tragen von bequemen orthopädischen Schuhen kann die Entwicklung früher Fußdeformitäten verhindern.
Die Patienten sollten einen Podologen, d. h. einen Fußspezialisten, aufsuchen, der frühzeitig trophische Weichteilveränderungen verhindern und gegebenenfalls geeignete Medikamente verschreiben kann.
Gehbehinderungen können durch das Tragen spezieller Orthesen (Knöchel-Fuß-Orthesen). Diese können die Beugung des Beins und des Unterschenkels auf der hinteren Seite kontrollieren, die Instabilität des Knöchels beseitigen und das Körpergleichgewicht verbessern.
Eine solche Vorrichtung ermöglicht es dem Patienten, sich ohne fremde Hilfe fortzubewegen, und verhindert unerwünschte Stürze und Verletzungen. Fußschienen werden bei einem Knickfußsyndrom eingesetzt.
Im Ausland gibt es ein gut entwickeltes System zur Unterstützung von Patienten und ihren Familien: ‚Eine Welt ohne die Charcot-Krankheit ohne Marie Toutat‘.
Es gibt verschiedene spezialisierte Organisationen, Gesellschaften und Stiftungen. Es wird ständig geforscht, um neue Behandlungsmöglichkeiten für diese Krankheit zu finden.
Leider gibt es in der Russischen Föderation keine derartigen Einrichtungen, aber die Forschung zur Erforschung und Entwicklung optimaler Behandlungsmethoden ist im Gange und recht aktiv.
Forschungsinstitute in Baschkortostan, Woronesch, Krasnojarsk, Nowokusnezk, Samara, Saratow und Tomsk führen solche Programme durch.
Lesen Sie mehr:- Was ist Charcot?.
- Röntgenbild des Charcot-Fußes.
- Was ist distal?.
- Marfan-Syndrom – Zusammenfassung.
- Parese des Unterkörpers.
- Schlurfen der Füße beim Gehen Ursachen.
- Syndrom des Nervus tibialis.
- Innervation der Unterschenkelmuskeln.